Citation: Dao-Peng Chen, Fangyuan Zhang, Shili Lin. AIJ: joint test for simultaneous detection of imprinting and non-imprinting allelic expression imbalance[J]. Mathematical Biosciences and Engineering, 2020, 17(1): 366-386. doi: 10.3934/mbe.2020020

| [1] | A. D. Goldberg, C. D. Allis and E. Bernstein, Epigenetics: a landscape takes shape, Cell, 128 (2007), 635-638. |
| [2] | A. C. Ferguson-Smith, Genomic imprinting: the emergence of an epigenetic paradigm, Nat. Rev. Genet., 12 (2011), 565-575. |
| [3] | I. M. Morison, C. J. Paton and S. D. Cleverley, The imprinted gene and parent-of-origin effect database, Nucleic Acids Res., 29 (2001), 275-276. |
| [4] | D. Haig, Evolutionary conflicts in pregnancy and calcium metabolism - a review, Placenta, 25 Suppl A (2004), S10-S15. |
| [5] | D. H. Lim and E. R. Maher, Human imprinting syndromes, Epigenomics, 1 (2009), 347-369. |
| [6] | X. Wang, Q. Sun, S. D. McGrath, et al., Transcriptome-wide identification of novel imprinted genes in neonatal mouse brain, PloS One, 3 (2008), e3839. |
| [7] | C. Gregg, J. W. Zhang, B. Weissbourd, et al., High-resolution analysis of parent-of-origin allelic expression in the mouse brain, Science, 329 (2010), 643-648. |
| [8] | Y. Takada, R. Miyagi, A. Takahashi, et al., A generalized linear model for decomposing cisregulatory, parent-of-origin, and maternal effects on allele-specific gene expression, G3-Genes Genom. Genet., 7 (2017), 2227-2234. |
| [9] | C. Wang, Z. Wang, D. R. Prows, et al., A computational framework for the inheritance pattern of genomic imprinting for complex traits, Brief. Bioinform., 13 (2012), 34-45. |
| [10] | T. He, J. Sa, P.-S. Zhong, et al., Statistical dissection of cyto-nuclear epistasis subject to genomic imprinting in line crosses, PloS One, 9 (2014), e91702. |
| [11] | T.-J. Chuang, Y.-H. Tseng, C.-Y. Chen, et al., Assessment of imprinting-and genetic variationdependent monoallelic expression using reciprocal allele descendants between human family trios, Sci. Rep., 7 (2017), 7038. |
| [12] | K. D. Falkenberg, N. E. Braverman, A. B. Moser, et al., Allelic expression imbalance promoting a mutant PEX6 allele causes Zellweger spectrum disorder, Am. J. Hum. Genet., 101 (2017), 965- 976. |
| [13] | P. Llavona, M. Pinelli, M. Mutarelli, et al., Allelic expression imbalance in the human retinal transcriptome and potential impact on inherited retinal diseases, Genes, 8 (2017), 283. |
| [14] | W. Lin, H.-D. Lin, X.-Y. Guo, et al., Allelic expression imbalance polymorphisms in susceptibility chromosome regions and the risk and survival of breast cancer, Mol. Carcinogen., 56 (2017), 300- 311. |
| [15] | J. M. Gahan, M. M. Byrne, E. Connolly, et al., 39 Allelic expression imbalance at interleukin 18 and chemokine cxcl 16 in patients with acute coronary syndromes, HEART, 101 (2015), A22, Annual Conference and AGM of the Irish-Cardiac-Society, Killarney, IRELAND, OCT 08-10, 2015. |
| [16] | L. B. Hesson, D. Packham, C.-T. Kwok, et al., Lynch syndrome associated with two MLH1 promoter variants and allelic imbalance of MLH1 expression, Hum. Mutat., 36 (2015), 622-630. |
| [17] | S. E. McCormack and S. F. A. Grant, Allelic expression imbalance: tipping the scales to elucidate the function of type 2 diabetes-associated loci, Diabetes, 64 (2015), 1102-1104. |
| [18] | A. K. Mondal, N. K. Sharma, S. C. Elbein, et al., Allelic expression imbalance screening of genes in chromosome 1q21-24 region to identify functional variants for Type 2 diabetes susceptibility, Physiol. Genomics, 45 (2013), 509-520. |
| [19] | J. Hu and I. Peter, Evidence of expression variation and allelic imbalance in Crohn's disease susceptibility genes NOD2 and ATG16L1 in human dendritic cells, Gene, 527 (2013), 496-502. |
| [20] | P. Fontanillas, C. R. Landry, P. J. Wittkopp, et al., Key considerations for measuring allelic expression on a genomic scale using high-throughput sequencing, Mol. Ecol., 19 Suppl 1 (2010), 212-227. |
| [21] | T. Goovaerts, S. Steyaert, C. A. Vandenbussche, et al., A comprehensive overview of genomic imprinting in breast and its deregulation in cancer, Nat. Commun., 9 (2018), 4120. |
| [22] | F. Zou, W. Sun, J. J. Crowley, et al., A novel statistical approach for jointly analyzing RNA-seq data from F-1 reciprocal crosses and inbred lines, Genetics, 197 (2014), 389-399. |
| [23] | J. Feng, C. A. Meyer, Q. Wang, et al., Gene expression GFOLD: a generalized fold change for ranking differentially expressed genes from RNA-seq data, Bioinformatics, 28 (2012), 2782-2788. |
| [24] | S. A. Seesi, Y. T. Tiagueu, A. Zelikovsky, et al., Bootstrap-based differential gene expression analysis for RNA-Seq data with and without replicates, BMC Genomics, 15 (2014), 1-10. |
| [25] | S. Tarazona, F. Garcia, J. Dopazo, et al., Differential expression in RNA-seq: A matter of depth, Genome Res., 2213-2223. |
| [26] | E. Topol, Individualized medicine from pre-womb to tomb, Cell, 157 (2014), 241-253. |
| [27] | N. J. Schork, Time for one-person trials, Nature, 520 (2015), 609-611. |
| [28] | J. Z. B. Low, T. F. Khang and M. T. Tammi, Open Access CORNAS: coverage-dependent RNA-Seq analysis of gene expression data without biological replicates, BMC Bioinformatics, 18 (2017), 575. |
| [29] | B. E. Storer and C. Kim, Exact properties of some exact test statistics for comparing two binomial proportions, J. Am. Stat. Assoc., 85 (1990), 146-155. |
| [30] | R. Little, Testing the equality of two independent binomial proportionsam stat, Am. Stat., 43 (1989), 283-288. |
| [31] | H. Luo and S. Lin, Evaluation of classical statistical methods for analyzing bs-seq data, OBM Genet., 2 (2018), 053. |
| [32] | M. Gehring, V. Missirian and S. Henikoff, Genomic analysis of parent-of-origin allelic expression in arabidopsis thaliana seeds, PloS One, 6 (2011), e23687. |
| [33] | D.-P. Chen, Statistical power for RNA-seq data to detect two epigenetic phenomena, Ph.D thesis, The Ohio State University, 2013. |
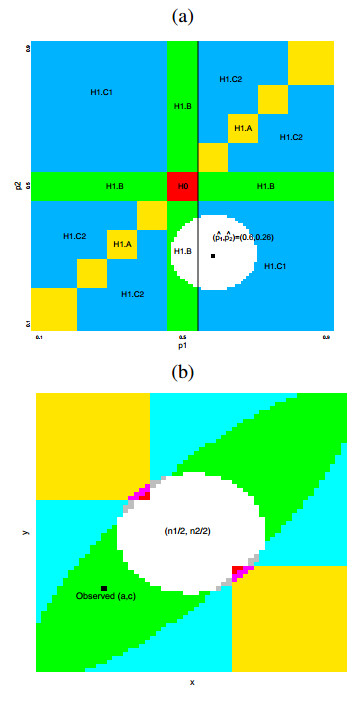
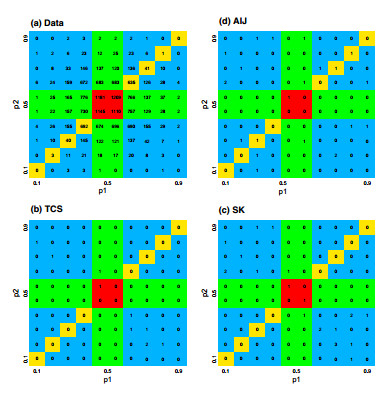
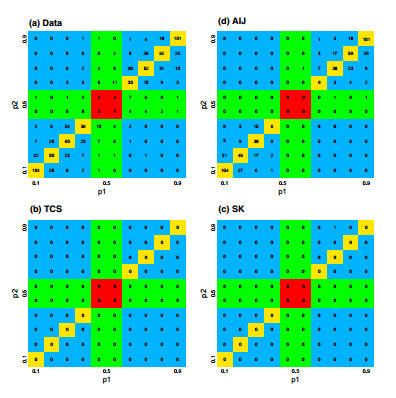
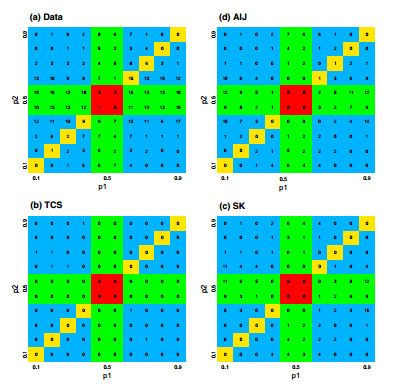
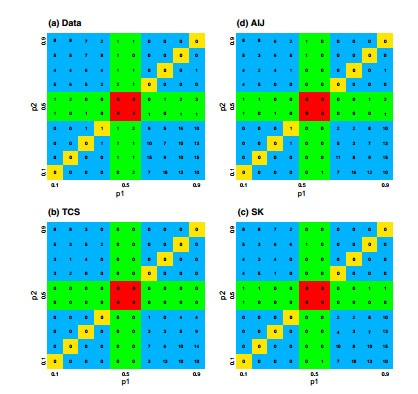
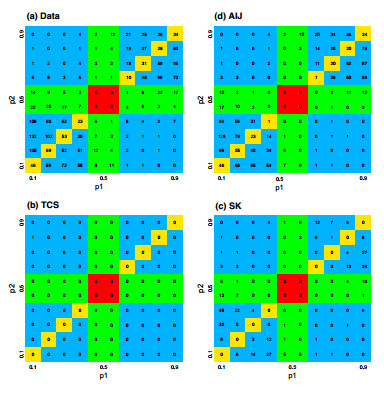
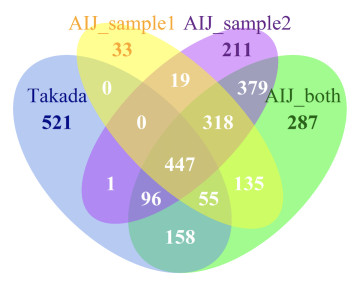
Figures(7) / Tables(5)

Dao-Peng Chen, Fangyuan Zhang, Shili Lin. AIJ: joint test for simultaneous detection of imprinting and non-imprinting allelic expression imbalance[J]. Mathematical Biosciences and Engineering, 2020, 17(1): 366-386. doi: 10.3934/mbe.2020020







 DownLoad:
DownLoad: