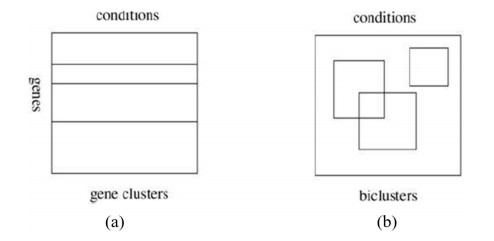
Microarray and RNA-sequencing (RNA-seq) techniques each produce gene expression data that can be expressed as a matrix that often contains missing values. Thus, a process of missing-value imputation that uses coherence information of the dataset is necessary. Existing imputation methods, such as iterative bicluster-based least squares (bi-iLS), use biclustering to estimate the missing values because genes are only similar under correlative experimental conditions. Also, they use the row average to obtain a temporary complete matrix, but the use of the row average is considered to be a flaw. The row average cannot reflect the real structure of the dataset because the row average only uses the information of an individual row. Therefore, we propose the use of Bayesian principal component analysis (BPCA) to obtain the temporary complete matrix instead of using the row average in bi-iLS. This alteration produces new missing values imputation method called iterative bicluster-based Bayesian principal component analysis and least squares (bi-BPCA-iLS). Several experiments have been conducted on two-dimension independent gene expression datasets, which are microarray (e.g., cell-cycle expression dataset of yeast saccharomyces cerevisiae) and RNA-seq (gene expression data from schizosaccharomyces pombe) datasets. In the case of the microarray dataset, our proposed bi-BPCA-iLS method showed a significant overall improvement in the normalized root mean square error (NRMSE) values of 10.6% from the local least squares (LLS) and 0.6% from the bi-iLS. In the case of the RNA-seq dataset, our proposed bi-BPCA-iLS method showed an overall improvement in the NRMSE values of 8.2% from the LLS and 3.1% from the bi-iLS. The additional computational time of bi-BPCA-iLS is not significant compared to bi-iLS.
Citation: Saskya Mary Soemartojo, Titin Siswantining, Yoel Fernando, Devvi Sarwinda, Herley Shaori Al-Ash, Sarah Syarofina, Noval Saputra. Iterative bicluster-based Bayesian principal component analysis and least squares for missing-value imputation in microarray and RNA-sequencing data[J]. Mathematical Biosciences and Engineering, 2022, 19(9): 8741-8759. doi: 10.3934/mbe.2022405
Microarray and RNA-sequencing (RNA-seq) techniques each produce gene expression data that can be expressed as a matrix that often contains missing values. Thus, a process of missing-value imputation that uses coherence information of the dataset is necessary. Existing imputation methods, such as iterative bicluster-based least squares (bi-iLS), use biclustering to estimate the missing values because genes are only similar under correlative experimental conditions. Also, they use the row average to obtain a temporary complete matrix, but the use of the row average is considered to be a flaw. The row average cannot reflect the real structure of the dataset because the row average only uses the information of an individual row. Therefore, we propose the use of Bayesian principal component analysis (BPCA) to obtain the temporary complete matrix instead of using the row average in bi-iLS. This alteration produces new missing values imputation method called iterative bicluster-based Bayesian principal component analysis and least squares (bi-BPCA-iLS). Several experiments have been conducted on two-dimension independent gene expression datasets, which are microarray (e.g., cell-cycle expression dataset of yeast saccharomyces cerevisiae) and RNA-seq (gene expression data from schizosaccharomyces pombe) datasets. In the case of the microarray dataset, our proposed bi-BPCA-iLS method showed a significant overall improvement in the normalized root mean square error (NRMSE) values of 10.6% from the local least squares (LLS) and 0.6% from the bi-iLS. In the case of the RNA-seq dataset, our proposed bi-BPCA-iLS method showed an overall improvement in the NRMSE values of 8.2% from the LLS and 3.1% from the bi-iLS. The additional computational time of bi-BPCA-iLS is not significant compared to bi-iLS.

| [1] |
T. Siswantining, A. Bustamam, S. Puspa, Z. Rustam, F. Zubedi, Biclustering of diabetic nephropathy and diabetic retinopathy microarray data using a similarity-based biclustering algorithm, Int. J. Bioinf. Res. Appl., 17 (2021), 343–362. https://doi.org/10.1504/ijbra.2021.117934 doi: 10.1504/ijbra.2021.117934

|
| [2] |
B. Pontes, R. Girldez, J. Aguilar-Ruiz, Quality measures for gene expression biclusters, PloS One, 10 (2015), e0115497. https://doi.org/10.1371/journal.pone.0115497 doi: 10.1371/journal.pone.0115497
|
| [3] |
S. Madeira, A. Oliveira, Biclustering algorithms for biological data analysis: A survey, IEEE/ACM Trans. Comput. Biol. Bioinf., 1 (2004), 24–45. https://doi.org/10.1109/TCBB.2004.2 doi: 10.1109/TCBB.2004.2
|
| [4] |
K. Cheng, N. Law, W. Siu, Iterative bicluster-based least square framework for estimation of missing values in microarray gene expression data, Pattern Recognit., 45 (2012), 1281–1289. https://doi.org/10.1016/j.patcog.2011.10.012 doi: 10.1016/j.patcog.2011.10.012
|
| [5] |
F. Shi, D. Zhang, J. Chen, H. Karimi, Missing value estimation for microarray data by Bayesian principal component analysis and iterative local least squares, Math. Prob. Eng., 2013 (2013), 1–5. https://doi.org/10.1155/2013/162938 doi: 10.1155/2013/162938
|
| [6] |
D. Rubin, Inference And missing data, Biometrika, 63 (1976), 581–592. https://doi.org/10.1093/biomet/63.3.581 doi: 10.1093/biomet/63.3.581
|
| [7] | S. Christopher, T. Siswantining, D. Sarwinda, A. Bustaman, Missing value analysis of numerical data using fractional hot deck imputation, in 2019 3rd International Conference On Informatics and Computational Sciences (ICICoS), (2019), 1–6. https://doi.org/10.1109/icicos48119.2019.8982412 |
| [8] |
A. G. De Brevern, S. Hazout, A. Malpertuy, Influence of microarrays experiments missing values on the stability of gene groups by hierarchical clustering, BMC Bioinf., 5 (2004), 1–12. https://doi.org/10.1186/1471-2105-5-114 doi: 10.1186/1471-2105-5-114
|
| [9] |
M. Celton, A. Malpertuy, G. Lelandais, A. G. De Brevern, Comparative analysis of missing value imputation methods to improve clustering and interpretation of microarray experiments, BMC Genomics, 11 (2010), 1–16. https://doi.org/10.1186/1471-2164-11-15 doi: 10.1186/1471-2164-11-15
|
| [10] |
T. Siswantining, T. Anwar, D. Sarwinda, H. Al-Ash, A novel centroid initialization in missing value imputation towards mixed datasets, Commun. Math. Biol. Neurosci., 11 (2021), 1–36. https://doi.org/10.28919/cmbn/5344 doi: 10.28919/cmbn/5344
|
| [11] | C. Mack, Z. Su, D. Weistreich, L. Research, Managing Missing Data in Patient Registries: Addendum to Registries for Evaluating Patient Outcomes: A User's Guide, Agency for Healthcare Research and Quality (US), 2018. |
| [12] | P. Berkhin, A survey of clustering data mining techniques, in Grouping Multidimensional Data, Springer, (2006), 25–71. https://doi.org/10.1007/3-540-28349-8_2 |
| [13] |
T. Siswantining, A. Aminanto, D. Sarwinda, O. Swasti, Biclustering analysis using plaid model on gene expression data of colon cancer, Austrian J. Stat., 50 (2021), 101–114. https://doi.org/10.17713/ajs.v50i5.1195 doi: 10.17713/ajs.v50i5.1195
|
| [14] |
H. Zhao, A. Liew, D. Wang, H. Yan, Biclustering analysis for pattern discovery: Current techniques, comparative studies and applications, Curr. Bioinf.. 7 (2012), 43–55. https://doi.org/10.2174/157489312799304413 doi: 10.2174/157489312799304413
|
| [15] |
A. Tanay, R. Sharan, R. Shamir, Biclustering algorithms: A survey. Handbook of computational molecular biology, 9 (2005), 122–124. https://doi.org/10.1201/9781420036275.ch26 doi: 10.1201/9781420036275.ch26
|
| [16] |
H. Kim, G. Golub, H. Park, Missing value estimation for DNA microarray gene expression data: Local least squares imputation, Bioinformatics, 21 (2004), 187–198. https://doi.org/10.1093/bioinformatics/bth499 doi: 10.1093/bioinformatics/bth499
|
| [17] |
T. H. Bø, B. Dysvik, I. Jonassen, LSimpute: Accurate estimation of missing values in microarray data with least squares methods, Nucleic Acids Res., 32 (2004), e34. https://doi.org/10.1093/nar/gnh026 doi: 10.1093/nar/gnh026
|
| [18] |
L. Bras, J. Menezes, Dealing with gene expression missing data, IEE Proc. Syst. Biol., 153 (2006), 105. https://doi.org/10.1049/ip-syb:20050056 doi: 10.1049/ip-syb:20050056
|
| [19] |
S. Oba, M. Sato, I. Takemasa, M. Monden, K. Matsubara, S. Ishii, A Bayesian missing value estimation method for gene expression profile data, Bioinformatics, 19 (2003), 2088–2096. https://doi.org/10.1093/bioinformatics/btg287 doi: 10.1093/bioinformatics/btg287
|
| [20] |
G. Brock, J. Shaffer, R. Blakesley, M. Lotz, G. Tseng, Which missing value imputation method to use in expression profiles: A comparative study and two selection schemes, BMC Bioinf., 9 (2008), 1–12. https://doi.org/10.1186/1471-2105-9-12 doi: 10.1186/1471-2105-9-12
|
| [21] |
O. Troyanskaya, M. Cantor, G. Sherlock, P. Brown, T. Hastie, R. Tibshirani, et al., Missing value estimation methods for DNA microarrays, Bioinformatics, 17 (2001), 520–525. https://doi.org/10.1093/bioinformatics/17.6.520 doi: 10.1093/bioinformatics/17.6.520
|
| [22] |
A. Bustamam, S. Formalidin, T. Siswantining, Z. Rustam, Finding correlated biclusters from microarray data using the modified lift algorithm based on new residue score, Int. J. Data Mining Bioinf., 24 (2020), 326. https://doi.org/10.1504/ijdmb.2020.113691 doi: 10.1504/ijdmb.2020.113691
|
| [23] |
P. Spellman, G. Sherlock, M. Zhang, V. Iyer, K. Anders, M. Eisen, et al., Comprehensive identification of cell cycle-regulated genes of the YeastSaccharomyces cerevisiaeby microarray hybridization, Mol. Biol. Cell, 9 (1998), 3273–3297. https://doi.org/10.1091/mbc.9.12.3273 doi: 10.1091/mbc.9.12.3273
|
| [24] |
C. Shan, C. Bao, J. Diedrich, X. Chen, C. Lu, J. Yates, et al., The INO80 complex regulates epigenetic inheritance of heterochromatin, Cell Rep., 33 (2020), 108561. https://doi.org/10.1016/j.celrep.2020.108561 doi: 10.1016/j.celrep.2020.108561
|








Figures(9) / Tables(9)

Saskya Mary Soemartojo, Titin Siswantining, Yoel Fernando, Devvi Sarwinda, Herley Shaori Al-Ash, Sarah Syarofina, Noval Saputra. Iterative bicluster-based Bayesian principal component analysis and least squares for missing-value imputation in microarray and RNA-sequencing data[J]. Mathematical Biosciences and Engineering, 2022, 19(9): 8741-8759. doi: 10.3934/mbe.2022405







 DownLoad:
DownLoad: