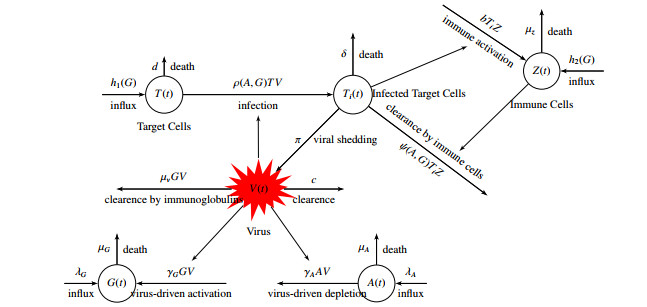
Human immunodeficiency virus (HIV) infection is a major public health concern with 1.2 million people living with HIV in the United States. The role of nutrition in general, and albumin/globulin in particular in HIV progression has long been recognized. However, no mathematical models exist to describe the interplay between HIV and albumin/globulin. In this paper, we present a family of models of HIV and the two protein components albumin and globulin. We use albumin, globulin, viral load and target cell data from simian immunodeficiency virus (SIV)-infected monkeys to perform model selection on the family of models. We discover that the simplest model accurately and uniquely describes the data. The selection of the simplest model leads to the observation that albumin and globulin do not impact the infection rate of target cells by the virus and the clearance of the infected target cells by the immune system. Moreover, the recruitment of target cells and immune cells are modeled independently of globulin in the selected model. Mathematical analysis of the selected model reveals that the model has an infection-free equilibrium and a unique infected equilibrium when the immunological reproduction number is above one. The infection-free equilibrium is locally stable when the immunological reproduction number is below one, and unstable when the immunological reproduction number is greater than one. The infection equilibrium is locally stable whenever it exists. To determine the parameters of the best fitted model we perform structural and practical identifiability analysis. The structural identifiability analysis reveals that the model is identifiable when the immune cell infection rate is fixed at a value obtained from the literature. Practical identifiability reveals that only seven of the sixteen parameters are practically identifiable with the given data. Practical identifiability of parameters performed with synthetic data sampled a lot more frequently reveals that only two parameters are practically unidentifiable. We conclude that experiments that will improve the quality of the data can help improve the parameter estimates and lead to better understanding of the interplay of HIV and albumin-globulin metabolism.
Citation: Vivek Sreejithkumar, Kia Ghods, Tharusha Bandara, Maia Martcheva, Necibe Tuncer. Modeling the interplay between albumin-globulin metabolism and HIV infection[J]. Mathematical Biosciences and Engineering, 2023, 20(11): 19527-19552. doi: 10.3934/mbe.2023865
Human immunodeficiency virus (HIV) infection is a major public health concern with 1.2 million people living with HIV in the United States. The role of nutrition in general, and albumin/globulin in particular in HIV progression has long been recognized. However, no mathematical models exist to describe the interplay between HIV and albumin/globulin. In this paper, we present a family of models of HIV and the two protein components albumin and globulin. We use albumin, globulin, viral load and target cell data from simian immunodeficiency virus (SIV)-infected monkeys to perform model selection on the family of models. We discover that the simplest model accurately and uniquely describes the data. The selection of the simplest model leads to the observation that albumin and globulin do not impact the infection rate of target cells by the virus and the clearance of the infected target cells by the immune system. Moreover, the recruitment of target cells and immune cells are modeled independently of globulin in the selected model. Mathematical analysis of the selected model reveals that the model has an infection-free equilibrium and a unique infected equilibrium when the immunological reproduction number is above one. The infection-free equilibrium is locally stable when the immunological reproduction number is below one, and unstable when the immunological reproduction number is greater than one. The infection equilibrium is locally stable whenever it exists. To determine the parameters of the best fitted model we perform structural and practical identifiability analysis. The structural identifiability analysis reveals that the model is identifiable when the immune cell infection rate is fixed at a value obtained from the literature. Practical identifiability reveals that only seven of the sixteen parameters are practically identifiable with the given data. Practical identifiability of parameters performed with synthetic data sampled a lot more frequently reveals that only two parameters are practically unidentifiable. We conclude that experiments that will improve the quality of the data can help improve the parameter estimates and lead to better understanding of the interplay of HIV and albumin-globulin metabolism.

| [1] | National Institute of Health, HIV and Nutrition and Food Safety, Accessed: 2023-01-12. Available from: https://hivinfo.nih.gov/understanding-hiv/fact-sheets/hiv-and-nutrition-and-food-safety. |
| [2] | N. S. Scrimshaw, J. P. SanGiovanni, Synergism of nutrition, infection and immunity: An overview, Am. J. Clin. Nutr., 66 (1997), 464S–477S. |
| [3] |
E. Colecraft, HIV/AIDS: nutritional implications and impact on human development, Proc. Nutr. Soc., 67 (1997), 109–113. https://doi.org/10.1017/S0029665108006095 doi: 10.1017/S0029665108006095

|
| [4] |
W. Baisel, Nutrition and immune function: Overview, J. Nutr., 126 (1996), 2611S–2615S. https://doi.org/10.1093/jn/126.suppl_10.2611S doi: 10.1093/jn/126.suppl_10.2611S
|
| [5] |
S. V. Thuppal, S. Jun, A. Cowan, R. L. Bailey, The nutritional status of HIV-infected US adults, Curr. Dev. Nutr., 1 (2017), 1–6. https://doi.org/10.3945/cdn.117.001636 doi: 10.3945/cdn.117.001636
|
| [6] | S. H. Mehta, J. Astemborski, T. R. Sterling, D. L. Thomas, D. Vlahov, Serum albumin as a prognostic indicator for HIV disease progression, AIDS Res. Hum. Retroviruses, 22 (2006), 14–21. https://doi.org/10.1089/aid.2006.22.14 |
| [7] | M. E. Linares, J. F. Bencomo, L. E. Perez, O. Torrez, O. Barrera, Influence of HIV/AIDS infection on some biochemical indicators of the nutritional status, Biomedica, 22 (2002), 116–122. |
| [8] |
S. H. Mehta, J. Astemborski, L. E. Perez, T. R. Sterling, D. L. Thomas, Serum albumin as a prognostic indicator for HIV disease progression, AIDS Res. Hum. Retroviruses, 22 (2006), 14–21. https://doi.org/10.1089/aid.2006.22.14 doi: 10.1089/aid.2006.22.14
|
| [9] |
L. Lang, Z. Zhu, Z. Xu, S. Zhu, P. Meng, H. Wang, et al., The association between the albumin and viral negative conversion rate in patients infected with novel coronavirus disease 2019 (COVID-19), Infect. Drug Resist., 15 (2022), 1687–1694. https://doi.org/10.2147/IDR.S353091 doi: 10.2147/IDR.S353091
|
| [10] |
N. Schenker, L. G. Borrud, V. L. Burt, L. R. Curtin, K. M. Flegal, J. Hughes, et al., Multiple imputation of missing dual-energy X-ray absorptiometry data in the National Health and Nutrition Examination Survey, Stat. Med., 30 (2011), 260–276. https://doi.org/10.1002/sim.4080 doi: 10.1002/sim.4080
|
| [11] |
S. P. Caudill, Use of pooled samples from the national health and nutrition examination survey, Stat. Med., 31 (2012), 3269–3277. https://doi.org/10.1002/sim.5341 doi: 10.1002/sim.5341
|
| [12] |
M. T. L. Vasconcellos, P. L. N. Silva, L. A. Anjos, Sample design for the nutrition, physical activity and health survey (PNAFS), Niterói, Rio de Janeiro, Brazil, Estadistica, 65 (2013), 83–98. https://doi.org/10.1007/978-3-319-43851-1 doi: 10.1007/978-3-319-43851-1
|
| [13] |
J. Ma, W. Chan, C. L. Tsai, M. Xiong, B. C. Tilley, Analysis of transtheoretical model of health behavioral changes in a nutrition intervention study—a continuous time Markov chain model with Bayesian approach, Stat. Med., 34 (2015), 3577–3589. https://doi.org/10.1002/sim.6571 doi: 10.1002/sim.6571
|
| [14] | J. A. Novotny, M. H. Green, R. C. Boston, Mathematical Modeling in Nutrition and the Health Sciences, in Advances in Experimental Medicine and Biology, Kluwer Academic/Plenum Publishers, New York, 2003. |
| [15] | B. Juillet, J. Salomon, D. Tomé, H. Fouillet, Development and calibration of a modeling tool for the analysis of clinical data in human nutrition, in ESAIM: Proceedings, EDP Sciences, 14 (2005), 124–155. https://doi.org/10.1051/proc: 2005011 |
| [16] |
D. A. Drew, A mathematical model for nutrient metabolic chemistry, Appl. Math., 9 (2018), 647–671. https://doi.org/10.4236/am.2018.96045 doi: 10.4236/am.2018.96045
|
| [17] |
M. Chudtong, A. De Gaetano, A mathematical model of food intake, Math. Biosci. Eng., 18 (2021), 1238–1279. https://doi.org/10.3934/mbe.2021067 doi: 10.3934/mbe.2021067
|
| [18] | N. H. Shah, F. A. Thakkar, B. M. Yeolekar, Optimal control model for poor nutrition in life cycle, Dyn. Contin. Discrete Impuls. Syst. Ser. B Appl. Algorithms, 24 (2017), 387–400. |
| [19] |
D. Baleanu, A. Jajarmi, E. Bonyah, M. Hajipour, New aspects of poor nutrition in the life cycle within the fractional calculus, Adv. Differ. Equations, 230 (2018), 14. https://doi.org/10.1186/s13662-018-1684-x doi: 10.1186/s13662-018-1684-x
|
| [20] | Mathematical Nutrition Models, Accessed: 2023-01-11. Available from: https://www.nutritionmodels.com. |
| [21] | S. Coburn, D. W. Townsend, Mathematical Modeling in Experimental Nutrition: Vitamins, Proteins, Methods, Academic Press, San Diego, 1996. |
| [22] | M. Nowak, R. May, Virus Dynamics: Mathematical Principles of Immunology and Virology, Oxford University Press, New York, USA, 2000. |
| [23] |
R. de Boer, A. Perelson, Target cell limited and immune control models of HIV infection: A comparison, J. Theor. Biol., 190 (1998), 201–214. https://doi.org/10.1006/jtbi.1997.0548 doi: 10.1006/jtbi.1997.0548
|
| [24] |
S. Baral, R. Antia, N. M. Dixit, A dynamical motif comprising the interactions between antigens and CD8 T cells may underlie the outcomes of viral infections, PNAS, 116 (2019), 17393–17398. https://doi.org/10.1073/pnas.1902178116 doi: 10.1073/pnas.1902178116
|
| [25] | SelfDecode, Albumin/Globulin Ratio: High and Low Ratios + Normal Range, Accessed: 2021-08-04. Available from: https://labs.selfdecode.com/blog/albumin-globulin-ratio/\#Normal_AlbuminGlobulin_Ratio. |
| [26] | J. W. Haefner, Modeling Biological Systems: Principles and Applications, 2nd edition, Springer, New York, 2005. |
| [27] | K. P. Burnham, D. R. Anderson, Model Selection and Multi Model Inference: A Practical Information-Theoretic Approach, Springer, 2002. |
| [28] | Healthline, Total Protein Test, Accessed: 2021-07-18. Available from: https://www.healthline.com/health/total-protein#results. |
| [29] | Y. S. Sarro, A. Tounkara, E. Tangara, O. Guindo, H. L. White, E. Chamot, et al., Serum protein electrophoresis: any role in monitoring for antiretroviral therapy, Afr. Health Sci., 10 (2010), 138–143. |
| [30] |
A. Ronit, S. Sharma, J. V. Baker, R. Mngqibisa, T. Delory, L. Caldeira, et al., Serum albumin as a prognostic marker for serious non-AIDS endpoints in the strategic timing of antiretroviral treatment (START) study, J. Infect. Dis., 217 (2017), 405–412. https://doi.org/10.1093/infdis/jix350 doi: 10.1093/infdis/jix350
|
| [31] |
J. M. Conway, A. S. Perelson, Post-treatment control of HIV infection, PNAS, 12 (2015), 5467–5472. https://doi.org/10.1073/pnas.1419162112 doi: 10.1073/pnas.1419162112
|
| [32] | S. Duggal, T. D. Chugh, A. K. Duggal, HIV and malnutrition: effects on immune system, Clin. Dev. Immunol., 2012 (2012). https://doi.org/10.1155/2012/784740 |
| [33] | N. K. Vaidya, R. M. Ribeiro, A. S. Perelson, A. Kumar, Modeling the effects of morphine on simian immunodeficiency virus dynamics, PLOS Comput. Biol., 12 (2016), https://doi.org/10.1371/journal.pcbi.1005127 |
| [34] | S. M. Graham, S. Holte, J. T. Kimata, M. H. Wener, J. Overbaugh, A decrease in Albumin in early SIV infection is related to viral pathogenicity, AIDS Res. Hum. Retroviruses, 25 (2009), 433–440. |
| [35] |
J. Sun, H. Li, J. Liu, J. Zhao, D. Yuan, J. Guo, et al., The DTI changes and peripheral blood test results corroborate the early brain damage of SIV-infected rhesus, Radiol. Infect. Dis., 6 (2019), 8–14. https://doi.org/10.1016/j.jrid.2019.01.001 doi: 10.1016/j.jrid.2019.01.001
|
| [36] |
L. Lavreys, J. M. Baeten, V. Chohan, R. S. McClelland, W. M. Hassan, B. A. Richardson, et al., Higher set point plasma viral load and more-severe acute HIV type 1 (HIV-1) illness predict mortality among high-risk HIV-1-infected african women, Clin. Infect. Dis., 42 (2006), 1333–1339. https://doi.org/10.1086/503258 doi: 10.1086/503258
|
| [37] |
J. M. Conway, A. S. Perelson, Post-treatment control of hiv infection, Proc. Natl. Acad. Sci., 112 (2015), 5467–5472. https://doi.org/10.1073/pnas.1419162112 doi: 10.1073/pnas.1419162112
|
| [38] |
H. Miao, X. Xia, A. S. Perelson, H. Wu, On identifiability of nonlinear ode models and applications in viral dynamics, SIAM Rev., 53 (2011), 3–39. https://doi.org/10.1137/090757009 doi: 10.1137/090757009
|
| [39] |
G. Bellu, M. Saccomani, S. Audoly, L. D'Angio, Daisy: a new software tool to test global identifiability of biological and physiological systems, Comput. Methods Programs Biomed., 88 (2007), 52–61. https://doi.org/10.1016/j.cmpb.2007.07.002 doi: 10.1016/j.cmpb.2007.07.002
|
| [40] |
N. Tuncer, C. Mohanakumar, S. Swanson, M. Martcheva, Efficacy of control measures in the control of Ebola, Liberia, 2014–2015, J. Biol. Dyn., 12 (2018), 913–937. https://doi.org/10.1080/17513758.2018.1535095 doi: 10.1080/17513758.2018.1535095
|
| [41] |
N. Tuncer, M. Martcheva, Determining reliable parameter estimates for within-host and within-vector models of zika virus, J. Biol. Dyn., 15 (2021), 430–454. https://doi.org/10.1080/17513758.2021.1970261 doi: 10.1080/17513758.2021.1970261
|



Figures(4) / Tables(6)

Vivek Sreejithkumar, Kia Ghods, Tharusha Bandara, Maia Martcheva, Necibe Tuncer. Modeling the interplay between albumin-globulin metabolism and HIV infection[J]. Mathematical Biosciences and Engineering, 2023, 20(11): 19527-19552. doi: 10.3934/mbe.2023865







 DownLoad:
DownLoad: