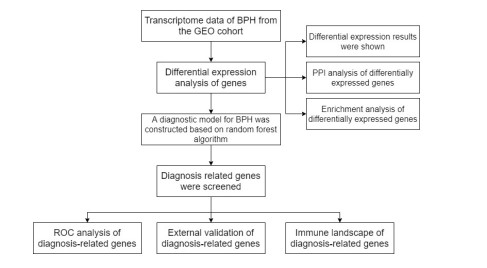
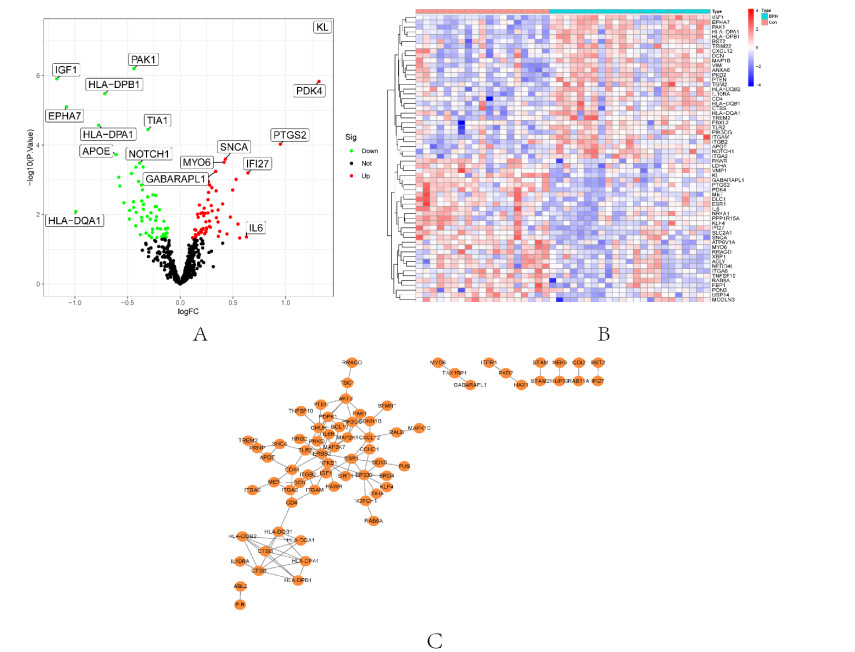
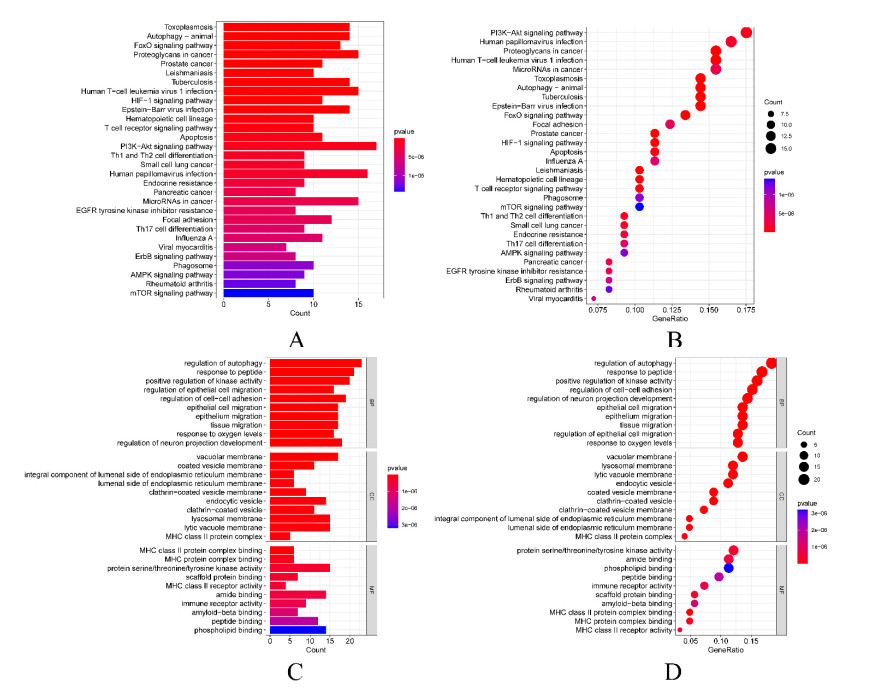
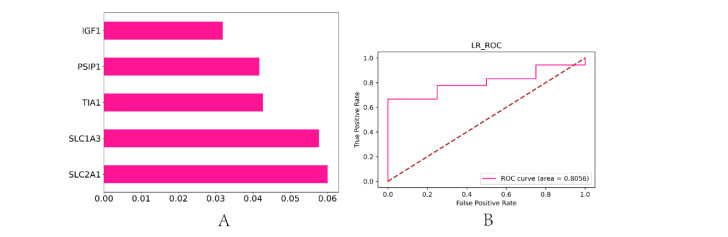
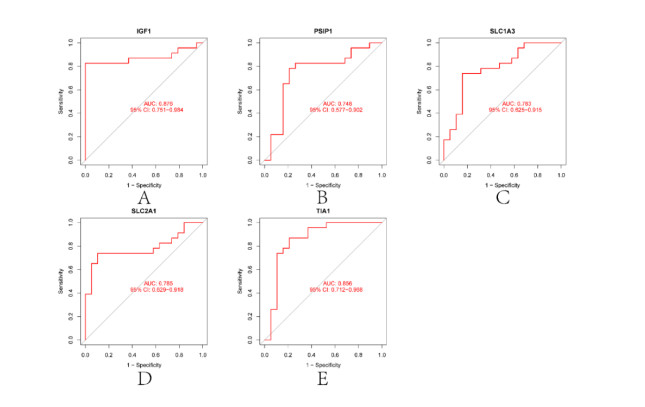
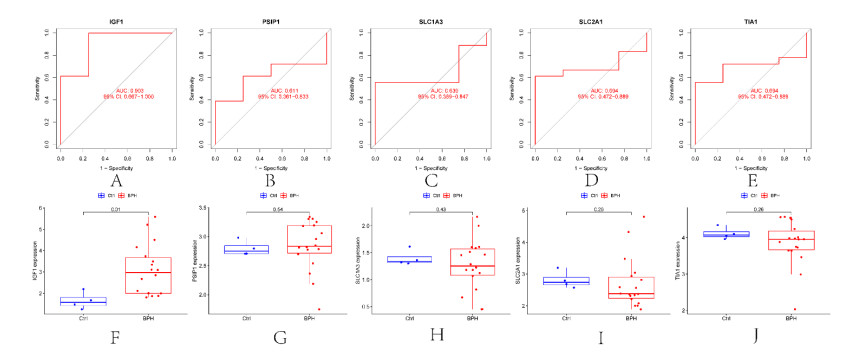
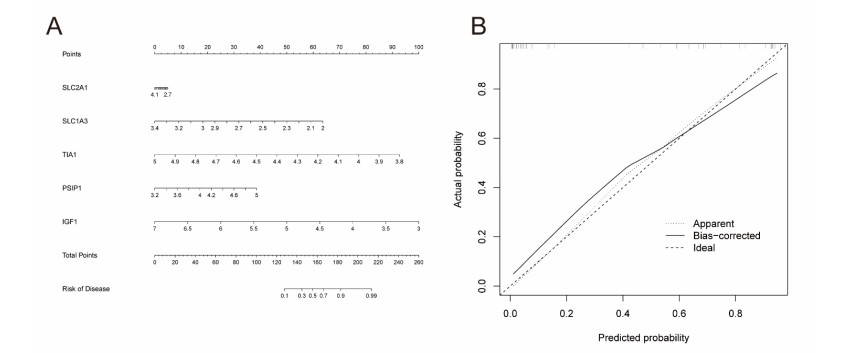
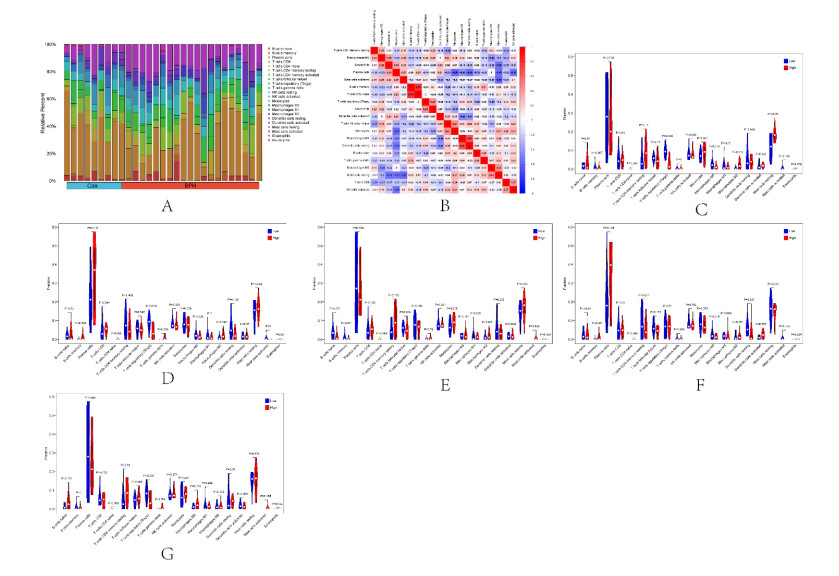
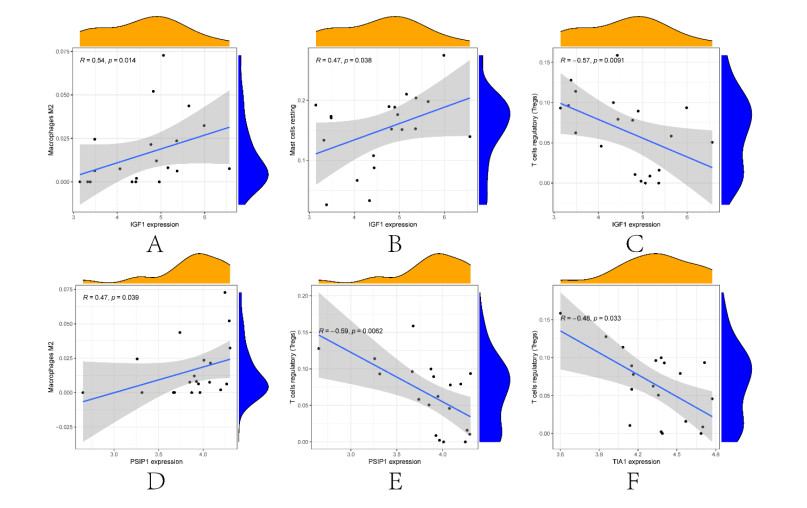
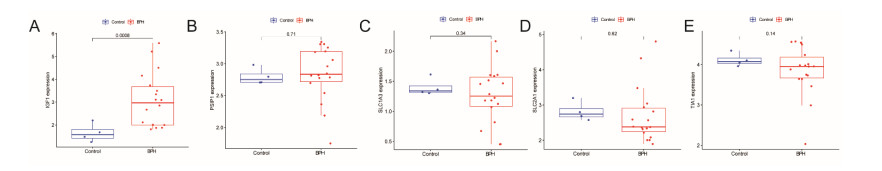
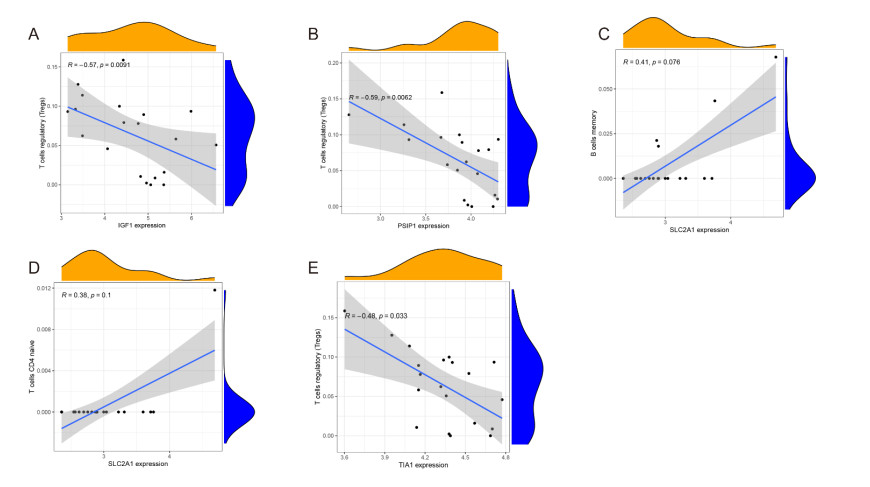
In older adults, benign prostatic hyperplasia (BPH) is the most common cause of lower urinary tract symptoms (LUTS). This study aimed to explore the genes with diagnostic value in patients with BPH, reveal the relationship between the expression of diagnosis-related genes and the immune microenvironment, and provide a reference for molecular diagnosis and immunotherapy of BPH. The combined gene expression data of GSE6099, GSE7307 and GSE119195 in the GEO database were used. The differential expression of autophagy-related genes between BPH patients and healthy controls was obtained by differential analysis. Then the genes related to BPH diagnosis were screened by a machine learning algorithm and verified. Finally, five important genes (IGF1, PSIP1, SLC1A3, SLC2A1 and T1A1) were obtained by random forest (RF) algorithm, and their relationships with the immune microenvironment were discussed. Five genes play an essential role in the occurrence and development of BPH and may become new diagnostic markers of BPH. Among them, immune cells have significant correlation with some genes. The signal transduction of IL-4 mediated by M2 macrophages is closely related to the progress of BPH. There are abundant active mast cells in BPH. The adoption and metastasis of regulatory T cells may be an important method to treat BPH.
Citation: Aiying Ying, Yueguang Zhao, Xiang Hu. Identification of biomarkers related to prostatic hyperplasia based on bioinformatics and machine learning[J]. Mathematical Biosciences and Engineering, 2023, 20(7): 12024-12038. doi: 10.3934/mbe.2023534
In older adults, benign prostatic hyperplasia (BPH) is the most common cause of lower urinary tract symptoms (LUTS). This study aimed to explore the genes with diagnostic value in patients with BPH, reveal the relationship between the expression of diagnosis-related genes and the immune microenvironment, and provide a reference for molecular diagnosis and immunotherapy of BPH. The combined gene expression data of GSE6099, GSE7307 and GSE119195 in the GEO database were used. The differential expression of autophagy-related genes between BPH patients and healthy controls was obtained by differential analysis. Then the genes related to BPH diagnosis were screened by a machine learning algorithm and verified. Finally, five important genes (IGF1, PSIP1, SLC1A3, SLC2A1 and T1A1) were obtained by random forest (RF) algorithm, and their relationships with the immune microenvironment were discussed. Five genes play an essential role in the occurrence and development of BPH and may become new diagnostic markers of BPH. Among them, immune cells have significant correlation with some genes. The signal transduction of IL-4 mediated by M2 macrophages is closely related to the progress of BPH. There are abundant active mast cells in BPH. The adoption and metastasis of regulatory T cells may be an important method to treat BPH.

| [1] |
H. Xiao, Y. Jiang, W. He, D. Xu, P. Chen, D. Liu, et al., Identification and functional activity of matrix-remodeling associated 5 (MXRA5) in benign hyperplastic prostate, Aging, 12 (2020), 8605–8621. https://doi.org/10.18632/aging.103175 doi: 10.18632/aging.103175

|
| [2] |
C. Wang, X. Du, R. Yang, J. Liu, D. Xu, J. Shi, et al., The prevention and treatment effects of tanshinone ⅡA on oestrogen/androgen-induced benign prostatic hyperplasia in rats, J. Steroid Biochem. Mol. Biol., 145 (2015), 28–37. https://doi.org/10.1016/j.jsbmb.2014.09.026 doi: 10.1016/j.jsbmb.2014.09.026
|
| [3] |
X. Li, Z. Dai, X. Wu, N. Zhang, H. Zhang, Z. Wang, et al., The comprehensive analysis identified an autophagy signature for the prognosis and the immunotherapy efficiency prediction in lung adenocarcinoma, Front. Immunol., 13 (2022), 749241. https://doi.org/10.3389/fimmu.2022.749241 doi: 10.3389/fimmu.2022.749241
|
| [4] |
S. He, Z. Deng, Z. Li, W. Gao, D. Zeng, Y. Shi, et al., Signatures of 4 autophagy-related genes as diagnostic markers of MDD and their correlation with immune infiltration, J. Affective Disord., 295 (2021), 11–20. https://doi.org/10.1016/j.jad.2021.08.005 doi: 10.1016/j.jad.2021.08.005
|
| [5] |
Z. Ke, H. Cai, Y. Wu, Y. Lin, X. Li, J. Huang, et al., Identification of key genes and pathways in benign prostatic hyperplasia, J. Cell. Physiol., 234 (2019), 19942–19950. https://doi.org/10.1002/jcp.28592 doi: 10.1002/jcp.28592
|
| [6] |
P. Xiang, D. Liu, D. Guan, Z. Du, Y. Hao, W. Yan, et al., Identification of key genes in benign prostatic hyperplasia using bioinformatics analysis, World J. Urol., 39 (2021), 3509–3516. https://doi.org/10.1007/s00345-021-03625-5 doi: 10.1007/s00345-021-03625-5
|
| [7] |
X. Xu, Y. Wang, Z. Sihong, J. Lu, X. Zheng, J. Wang, et al., Immune infiltration pattern associated with diagnosis and development in benign prostatic hyperplasia, Urol. J., 18 (2021), 564–572. https://doi.org/10.22037/uj.v18i.6678 doi: 10.22037/uj.v18i.6678
|
| [8] |
R. Sachdeva, N. Kaur, P. Kapoor, P. Singla, N. Thakur, S. Singhmar, Computational analysis of protein-protein interaction network of differentially expressed genes in benign prostatic hyperplasia, Mol. Biol. Res. Commun., 11 (2022), 85–96. https://doi.org/10.22099/mbrc.2022.43721.1746 doi: 10.22099/mbrc.2022.43721.1746
|
| [9] |
Y. Ge, Q. Wang, W. Shao, Y. Zhao, Q. Shi, Q. Yuan, et al., Circulating let-7f-5p improve risk prediction of prostate cancer in patients with benign prostatic hyperplasia, J. Cancer, 11 (2020), 4542–4549. https://doi.org/10.7150/jca.45077 doi: 10.7150/jca.45077
|
| [10] |
M. A. Harris, J. Clark, A. Ireland, J. Lomax, M. Ashburner, R. Foulger, et al., The Gene Ontology (GO) database and informatics resource, Nucleic Acids Res., 32 (2004), 258–261. https://doi.org/10.1093/nar/gkh036 doi: 10.1093/nar/gkh036
|
| [11] |
H. Ogata, S. Goto, K. Sato, W. Fujibuchi, H. Bono, M. Kanehisa, KEGG: Kyoto encyclopedia of genes and genomes, Nucleic Acids Res., 27 (1999), 29–34. https://doi.org/10.1093/nar/27.1.29 doi: 10.1093/nar/27.1.29
|
| [12] |
A. M. Newman, C. B. Steen, C. L. Liu, A. J. Gentles, A. A. Chaudhuri, F. Scherer, et al., Determining cell type abundance and expression from bulk tissues with digital cytometry, Nat. Biotechnol., 37 (2019), 773–782. https://doi.org/10.1038/s41587-019-0114-2 doi: 10.1038/s41587-019-0114-2
|
| [13] |
S. Wang, C. Zhang, Z. Xu, M. H. Chen, H. Yu, L. Wang, et al., Differential impact of PI3K/AKT/mTOR signaling on tumor initiation and progression in animal models of prostate cancer, Prostate, 83 (2023), 97–108. https://doi.org/10.1002/pros.24441 doi: 10.1002/pros.24441
|
| [14] |
S. Wang, K. Li, Z. Liu, S. Gui, N. Liu, X. Liu, Aerobic exercise ameliorates benign prostatic hyperplasia in obese mice through downregulating the AR/androgen/PI3K/AKT signaling pathway, Exp. Gerontol., 143 (2021), 111152. https://doi.org/10.1016/j.exger.2020.111152 doi: 10.1016/j.exger.2020.111152
|
| [15] |
G. J. Leiros, S. R. Galliano, M. E. Sember, T. Kahn, E. Schwarz, K. Eiguchi, Detection of human papillomavirus DNA and p53 codon 72 polymorphism in prostate carcinomas of patients from Argentina, BMC Urol., 5 (2005). https://doi.org/10.1186/1471-2490-5-15 doi: 10.1186/1471-2490-5-15
|
| [16] |
D. B. Joseph, G. H. Henry, A. Malewska, J. C. Reese, R. J. Mauck, J. C. Gahan, et al., 5-Alpha reductase inhibitors induce a prostate luminal to club cell transition in human benign prostatic hyperplasia, J. Pathol., 256 (2022), 427–441. https://doi.org/10.1002/path.5857 doi: 10.1002/path.5857
|
| [17] |
Y. Li, Q. Wang, J. Li, B. Shi, Y. Liu, P. Wang, SIRT3 affects mitochondrial metabolic reprogramming via the AMPK-PGC-1α axis in the development of benign prostatic hyperplasia, Prostate, 81 (2021), 1135–1148. https://doi.org/10.1002/pros.24208 doi: 10.1002/pros.24208
|
| [18] |
R. Liu, S. Zhang, R. Wan, J. Deng, W. Fang, Effect of Beclin-1 gene silencing on autophagy and apoptosis of the prostatic hyperplasia epithelial cells, Clinics, 77 (2022), 100076. https://doi.org/10.1016/j.clinsp.2022.100076 doi: 10.1016/j.clinsp.2022.100076
|
| [19] |
T. Kunit, C. Gratzke, A. Schreiber, F. Strittmatter, R. Waidelich, B. Rutz, et al., Inhibition of smooth muscle force generation by focal adhesion kinase inhibitors in the hyperplastic human prostate, Am. J. Physiol. Renal. Physiol., 307 (2014), 823–832. https://doi.org/10.1152/ajprenal.00011.2014 doi: 10.1152/ajprenal.00011.2014
|
| [20] |
N. Di, N. Mao, W. Cheng, H. Pang, Y. Ren, N. Wang, et al., Blood oxygenation level-dependent magnetic resonance imaging during carbogen breathing: differentiation between prostate cancer and benign prostate hyperplasia and correlation with vessel maturity, Onco Targets Ther., 9 (2016), 4143–4150. https://doi.org/10.2147/OTT.S105480 doi: 10.2147/OTT.S105480
|
| [21] |
G. Penna, B. Fibbi, S. Amuchastegui, C. Cossetti, F. Aquilano, G. Laverny, et al., Human benign prostatic hyperplasia stromal cells as inducers and targets of chronic immuno-mediated inflammation, J. Immunol., 182 (2009), 4056–4064. https://doi.org/10.4049/jimmunol.0801875 doi: 10.4049/jimmunol.0801875
|
| [22] |
U. Lehnigk, U. Zimmermann, C. Woenckhaus, J. Giebel, Localization of annexins Ⅰ, Ⅱ, Ⅳ and Ⅶ in whole prostate sections from radical prostatectomy patients, Histol. Histopathol., 20 (2005), 673–680. https://doi.org/10.14670/HH-20.673 doi: 10.14670/HH-20.673
|
| [23] | N. Soulitzis, I. Karyotis, D. Delakas, D. A. Spandidos, Expression analysis of peptide growth factors VEGF, FGF2, TGFB1, EGF and IGF1 in prostate cancer and benign prostatic hyperplasia, Int. J. Oncol., 29 (2006), 305–314. |
| [24] |
D. J. O'Rourke, D. A. DiJohnson, R. J. Caiazzo Jr, J. C. Nelson, D. Ure, M. P. O'Leary, et al. Autoantibody signatures as biomarkers to distinguish prostate cancer from benign prostatic hyperplasia in patients with increased serum prostate specific antigen, Clin. Chim. Acta, 413 (2012): 561–567. https://doi.org/10.1016/j.cca.2011.11.027 doi: 10.1016/j.cca.2011.11.027
|
| [25] |
J. Sheng, Y. Yang, Y. Cui, S. He, L. Wang, L. Liu, et al., M2 macrophage-mediated interleukin-4 signalling induces myofibroblast phenotype during the progression of benign prostatic hyperplasia, Cell Death Dis., 9 (2018), 755. https://doi.org/10.1038/s41419-018-0744-1 doi: 10.1038/s41419-018-0744-1
|
| [26] |
X. Jin, T. Lin, G. Yang, H. Cai, B. Tang, X. Liao, et al. Use of Tregs as a cell-based therapy via CD39 for benign prostate hyperplasia with inflammation, J. Cell. Mol. Med., 24 (2020), 5082–5096. https://doi.org/10.1111/jcmm.15137 doi: 10.1111/jcmm.15137
|
| [27] |
C. Wang, C. Han, Q. Zhao, X. Chen, Circular RNAs and complex diseases: from experimental results to computational models, Brief Bioinform, 22 (2021), 286. https://doi.org/10.1093/bib/bbab286 doi: 10.1093/bib/bbab286
|
| [28] |
W. Wang, L. Zhang, J. Sun, Q. Zhao, J. Shuai, Predicting the potential human lncRNA-miRNA interactions based on graph convolution network with conditional random field, Brief. Bioinform., 23 (2022), 463. https://doi.org/10.1093/bib/bbac463 doi: 10.1093/bib/bbac463
|
| [29] |
F. Sun, J. Sun, Q. Zhao, A deep learning method for predicting metabolite-disease associations via graph neural network, Brief. Bioinform., 23 (2022), 266. https://doi.org/10.1093/bib/bbac266 doi: 10.1093/bib/bbac266
|
| [30] |
L. Zhang, P. Yang, H. Feng, Q. Zhao, H. Liu, Using network distance analysis to predict lncRNA-miRNA interactions, Interdiscip Sci, 3 (2021), 535–545. https://doi.org/10.1007/s12539-021-00458-z doi: 10.1007/s12539-021-00458-z
|
Figures(11)

Aiying Ying, Yueguang Zhao, Xiang Hu. Identification of biomarkers related to prostatic hyperplasia based on bioinformatics and machine learning[J]. Mathematical Biosciences and Engineering, 2023, 20(7): 12024-12038. doi: 10.3934/mbe.2023534







 DownLoad:
DownLoad: