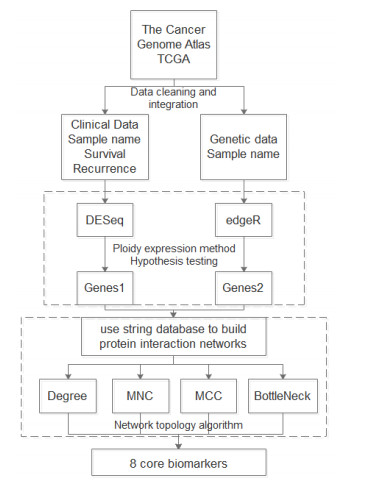
Liver cancer is a common cause of death from cancer in the population, with the 4th highest mortality rate from cancer worldwide. The high recurrence rate of hepatocellular carcinoma after surgery is an important cause of high mortality among patients. In this paper, based on eight scheduled core markers of liver cancer, an improved feature screening algorithm was proposed based on the analysis of the basic principles of the random forest algorithm, and the system was finally applied to liver cancer prognosis prediction to improve the prediction of biomarkers for liver cancer recurrence, and the impact of different algorithmic strategies on the prediction accuracy was compared and analyzed. The results showed that the improved feature screening algorithm was able to reduce the feature set by about 50% while ensuring that the prediction accuracy was reduced within 2%.
Citation: Zhiyue Su, Chengquan Li, Haitian Fu, Liyang Wang, Meilong Wu, Xiaobin Feng. Improved prognostic prediction model for liver cancer based on biomarker data screened by combined methods[J]. Mathematical Biosciences and Engineering, 2023, 20(3): 5316-5332. doi: 10.3934/mbe.2023246
Liver cancer is a common cause of death from cancer in the population, with the 4th highest mortality rate from cancer worldwide. The high recurrence rate of hepatocellular carcinoma after surgery is an important cause of high mortality among patients. In this paper, based on eight scheduled core markers of liver cancer, an improved feature screening algorithm was proposed based on the analysis of the basic principles of the random forest algorithm, and the system was finally applied to liver cancer prognosis prediction to improve the prediction of biomarkers for liver cancer recurrence, and the impact of different algorithmic strategies on the prediction accuracy was compared and analyzed. The results showed that the improved feature screening algorithm was able to reduce the feature set by about 50% while ensuring that the prediction accuracy was reduced within 2%.

| [1] | Cancer International Agency for Research on Cancer, Cancer Today, 2020. Available from: https://gco.iarc.fr/today/home. |
| [2] |
Z. Obermeyer, E. J. Emanuel, Predicting the future—Big data, machine learning, and clinical medicine, N. Engl. J. Med., 375 (2016), 1216. https://doi.org/10.1056/NEJMp1606181 doi: 10.1056/NEJMp1606181

|
| [3] |
V. K. Dhiman, M. J. Bolt, K. P. White, Nuclear receptors in cancer—Uncovering new and evolving roles through genomic analysis, Nat. Rev. Genet., 19 (2018), 160–174. https://doi.org/10.1038/nrg.2017.102 doi: 10.1038/nrg.2017.102
|
| [4] |
Z. Zhang, The role of big-data in clinical studies in laboratory medicine, J. Lab. Precis. Med., 2 (2017), 34. https://doi.org/10.21037/jlpm.2017.06.07 doi: 10.21037/jlpm.2017.06.07
|
| [5] | P. Han, R. Chu, C. C. Zhang, X. Guo, Clinical value of AFP-L3 in the diagnosis of hepatocellular carcinoma, Chin. J. Mod. Med., 14 (2012), 26–27. |
| [6] |
J. D. Yang, I. Nakamura, L. R. Roberts, The tumor microenvironment in hepatocellular carcinoma: Current status and therapeutic targets, Semin. Cancer Biol., 21 (2011), 35–43. https://doi.org/10.1016/j.semcancer.2010.10.007 doi: 10.1016/j.semcancer.2010.10.007
|
| [7] |
M. Hao, K. Liu, X. Guo, Y. Ouyang, D. Liu, D. Chen, et al., Effect of transforming growth factor-β on stem cell development and recurrence of hepatocellular carcinoma after radiofrequency ablation therapy, Beijing Med., 2016, 38 (2016), 1290–1294. https://doi.org/10.15932/j.0253-9713.2016.12.010 doi: 10.15932/j.0253-9713.2016.12.010
|
| [8] |
L. Jiang, Q. Yan, S. Fang, M. Liu, Y. Li, Y. F. Yuan, et al., Calcium-binding protein 39 promotes hepatocellular carcinoma growth and metastasis by activating extracellular signal-regulated kinase signaling pathway, Hepatology, 66 (2017), 1529–1545. https://doi.org/10.1002/hep.29312 doi: 10.1002/hep.29312
|
| [9] |
S. Umeda, M. Kanda, H. Sugimoto, H. Tanaka, M. Hayashi, S. Yamada, et al., Downregulation of GPR155 as a prognostic factor after curative resection of hepatocellular carcinoma, BMC Cancer, 17 (2017), 1–8. https://doi.org/10.1186/s12885-017-3629-2 doi: 10.1186/s12885-017-3629-2
|
| [10] |
S. Roessler, H. L. Jia, A. Budhu, M. Forgues, Q. H. Ye, J. S. Lee, et al., A unique metastasis gene signature enables prediction of tumor relapse in early-stage hepatocellular carcinoma patients, Cancer Res., 70 (2010), 10202–10212. https://doi.org/10.1158/0008-5472.CAN-10-2607 doi: 10.1158/0008-5472.CAN-10-2607
|
| [11] |
R. Genuer, J. M. Poggi, C. Tuleau-Malot, Variable selection using random forests, Pattern Recogn. Lett., 31 (2010), 2225–2236. https://doi.org/10.1016/j.patrec.2010.03.014 doi: 10.1016/j.patrec.2010.03.014
|
| [12] | D. Yao, J. Yang, X. Zhan, Feature selection algorithm based on random forest, J. Jilin Univ., 44 (2014), 137–141. |
| [13] | Matplotlib Developers, Annotations 2.2.2, 2012. Available from: https://matplotlib.org/2.2.2/tutorials/text/annotations.html. |








Figures(9) / Tables(8)

Zhiyue Su, Chengquan Li, Haitian Fu, Liyang Wang, Meilong Wu, Xiaobin Feng. Improved prognostic prediction model for liver cancer based on biomarker data screened by combined methods[J]. Mathematical Biosciences and Engineering, 2023, 20(3): 5316-5332. doi: 10.3934/mbe.2023246







 DownLoad:
DownLoad: