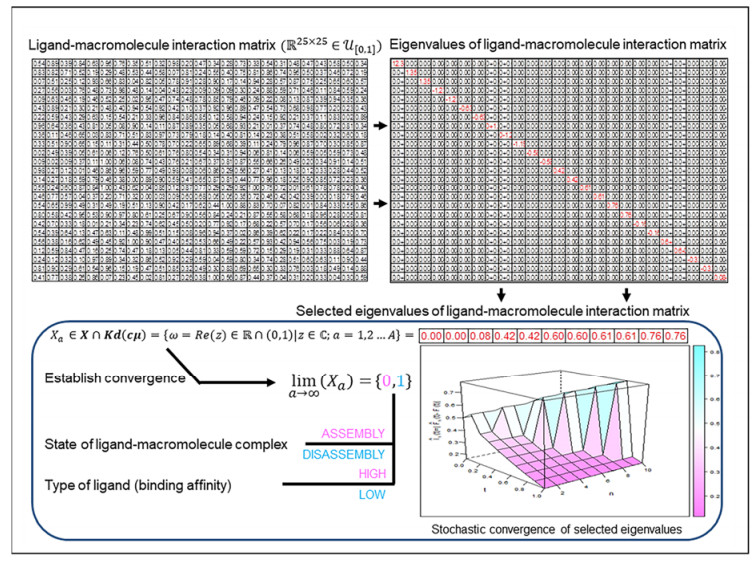
A ligand when bound to a macromolecule (protein, DNA, RNA) will influence the biochemical function of that macromolecule. This observation is empirical and attributable to the association of the ligand with the amino acids/nucleotides that comprise the macromolecule. The binding affinity is a measure of the strength-of-association of a macromolecule for its ligand and is numerically characterized by the association/dissociation constant. However, despite being widely used, a mathematically rigorous explanation by which the association/dissociation constant can influence the biochemistry and molecular biology of the resulting complex is not available. Here, the ligand-macromolecular complex is modeled as a homo- or hetero-dimer with a finite and equal number of atoms/residues per monomer. The pairwise interactions are numeric, empirically motivated and are randomly chosen from a standard uniform distribution. The transition-state dissociation constants are the strictly positive real part of all complex eigenvalues of this interaction matrix, belong to the open interval $(0, 1)$, and form a sequence whose terms are finite, monotonic, non-increasing and convergent. The theoretical results are rigorous, presented as theorems, lemmas and corollaries and are complemented by numerical studies. An inferential analysis of the clinical outcomes of amino acid substitutions of selected enzyme homodimers is also presented. These findings are extendible to higher-order complexes such as those likely to occur in vivo. The study also presents a schema by which a ligand can be annotated and partitioned into high- and low-affinity variants. The influence of the transition-state dissociation constants on the biochemistry and molecular biology of non-haem iron (Ⅱ)- and 2-oxoglutarate-dependent dioxygenases (catalysis) and major histocompatibility complex (Ⅰ) mediated export of high-affinity peptides (non-enzymatic association/dissociation) are examined as special cases.
Citation: Siddhartha Kundu. Modeling ligand-macromolecular interactions as eigenvalue-based transition-state dissociation constants may offer insights into biochemical function of the resulting complexes[J]. Mathematical Biosciences and Engineering, 2022, 19(12): 13252-13275. doi: 10.3934/mbe.2022620
A ligand when bound to a macromolecule (protein, DNA, RNA) will influence the biochemical function of that macromolecule. This observation is empirical and attributable to the association of the ligand with the amino acids/nucleotides that comprise the macromolecule. The binding affinity is a measure of the strength-of-association of a macromolecule for its ligand and is numerically characterized by the association/dissociation constant. However, despite being widely used, a mathematically rigorous explanation by which the association/dissociation constant can influence the biochemistry and molecular biology of the resulting complex is not available. Here, the ligand-macromolecular complex is modeled as a homo- or hetero-dimer with a finite and equal number of atoms/residues per monomer. The pairwise interactions are numeric, empirically motivated and are randomly chosen from a standard uniform distribution. The transition-state dissociation constants are the strictly positive real part of all complex eigenvalues of this interaction matrix, belong to the open interval $(0, 1)$, and form a sequence whose terms are finite, monotonic, non-increasing and convergent. The theoretical results are rigorous, presented as theorems, lemmas and corollaries and are complemented by numerical studies. An inferential analysis of the clinical outcomes of amino acid substitutions of selected enzyme homodimers is also presented. These findings are extendible to higher-order complexes such as those likely to occur in vivo. The study also presents a schema by which a ligand can be annotated and partitioned into high- and low-affinity variants. The influence of the transition-state dissociation constants on the biochemistry and molecular biology of non-haem iron (Ⅱ)- and 2-oxoglutarate-dependent dioxygenases (catalysis) and major histocompatibility complex (Ⅰ) mediated export of high-affinity peptides (non-enzymatic association/dissociation) are examined as special cases.

| [1] |
M. Su, Y. Ling, J. Yu, J. Wu, J. Xiao, Small proteins: untapped area of potential biological importance, Front. Genet., 4 (2013), 286. https://doi.org/10.3389/fgene.2013.00286 doi: 10.3389/fgene.2013.00286

|
| [2] |
M. B. Pappalardi, D. E. McNulty, J. D. Martin, K. E. Fisher, Y. Jiang, M. C. Burns, et al., Biochemical characterization of human HIF hydroxylases using HIF protein substrates that contain all three hydroxylation sites, Biochem. J., 436 (2011), 363–369. https://doi.org/10.1042/BJ20101201 doi: 10.1042/BJ20101201
|
| [3] | L. Esposito, M. Ferrara, L. Tomasi, P. De Filippo, Hereditary methemoglobinemia caused by NADH methemoglobin reductase deficiency, Pediatria (Napoli), 84 (1976), 411–422. |
| [4] |
D. E. Koshland Jr., G. Nemethy, D. Filmer, Comparison of experimental binding data and theoretical models in proteins containing subunits, Biochemistry, 5 (1966), 365–385. https://doi.org/10.1021/bi00865a047 doi: 10.1021/bi00865a047
|
| [5] |
J. Monod, J. Wyman, J. P. Changeux, On the nature of allosteric transitions: A plausible model, J. Mol. Biol., 12 (1965), 88–118. https://doi.org/10.1016/S0022-2836(65)80285-6 doi: 10.1016/S0022-2836(65)80285-6
|
| [6] |
J. J. Hutton Jr., A. L. Trappel, S. Udenfriend, Requirements for alpha-ketoglutarate, ferrous ion and ascorbate by collagen proline hydroxylase, Biochem. Biophys. Res. Commun., 24 (1966), 179–184. https://doi.org/10.1016/0006-291X(66)90716-9 doi: 10.1016/0006-291X(66)90716-9
|
| [7] |
S. Pektas, C. Y. Taabazuing, M. J. Knapp, Increased turnover at limiting O2 concentrations by the Thr387 → Ala variant of HIF-Prolyl Hydroxylase PHD2, Biochemistry, 54 (2015), 2851–2857. https://doi.org/10.1021/bi501540c doi: 10.1021/bi501540c
|
| [8] |
K. S. Hewitson, B. M. Lienard, M. A. McDonough, I. J. Clifton, D. Butler, A. S. Soares, et al., Structural and mechanistic studies on the inhibition of the hypoxia-inducible transcription factor hydroxylases by tricarboxylic acid cycle intermediates, J. Biol. Chem., 282 (2007), 3293–301. https://doi.org/10.1074/jbc.M608337200 doi: 10.1074/jbc.M608337200
|
| [9] |
K. M. Paulsson, M. J. Kleijmeer, J. Griffith, M. Jevon, S. Chen, P. O. Anderson, et al., Association of tapasin and COPI provides a mechanism for the retrograde transport of major histocompatibility complex (MHC) class Ⅰ molecules from the Golgi complex to the endoplasmic reticulum, J. Biol. Chem., 277 (2002), 18266–18271. https://doi.org/10.1074/jbc.M201388200 doi: 10.1074/jbc.M201388200
|
| [10] |
R. Benesch, R. E. Benesch, The effect of organic phosphates from the human erythrocyte on the allosteric properties of haemoglobin, Biochem. Biophys. Res. Commun., 26 (1967), 162–167. https://doi.org/10.1016/0006-291X(67)90228-8 doi: 10.1016/0006-291X(67)90228-8
|
| [11] |
P. J. Mulquiney, W. A. Bubb, P. W. Kuchel, Model of 2, 3-bisphosphoglycerate metabolism in the human erythrocyte based on detailed enzyme kinetic equations: in vivo kinetic characterization of 2, 3-bisphosphoglycerate synthase/phosphatase using 13C and 31P NMR, Biochem. J., 342 (1999), 567–580. https://doi.org/10.1042/bj3420567 doi: 10.1042/bj3420567
|
| [12] |
S. Martinez, R. P. Hausinger, Catalytic Mechanisms of Fe(Ⅱ)- and 2-Oxoglutarate-dependent Oxygenases, J. Biol. Chem., 290 (2015), 20702–20711. https://doi.org/10.1074/jbc.R115.648691 doi: 10.1074/jbc.R115.648691
|
| [13] |
I. J. Clifton, M. A. McDonough, D. Ehrismann, N. J. Kershaw, N. Granatino, C. J. Schofield, Structural studies on 2-oxoglutarate oxygenases and related double-stranded beta-helix fold proteins, J Inorg Biochem., 100 (2006), 644–669. https://doi.org/10.1016/j.jinorgbio.2006.01.024 doi: 10.1016/j.jinorgbio.2006.01.024
|
| [14] |
K. L. Gorres, R. T. Raines, Prolyl 4-hydroxylase, Crit. Rev. Biochem. Mol. Biol., 45 (2010), 106–124. https://doi.org/10.3109/10409231003627991 doi: 10.3109/10409231003627991
|
| [15] |
E. Hausmann, Cofactor requirements for the enzymatic hydroxylation of lysine in a polypeptide precursor of collagen, Biochim. Biophys. Acta, Protein Struct., 133 (1967), 591–593. https://doi.org/10.1016/0005-2795(67)90566-1 doi: 10.1016/0005-2795(67)90566-1
|
| [16] |
M. J. Landrum, J. M. Lee, M. Benson, G. R. Brown, C. Chao, S. Chitipiralla, et al., ClinVar: improving access to variant interpretations and supporting evidence, Nucleic Acids Res., 46 (2018), D1062–D1067. https://doi.org/10.1093/nar/gkx1153 doi: 10.1093/nar/gkx1153
|
| [17] |
S. Richards, N. Aziz, S. Bale, D. Bick, S. Das, J. Gastier-Foster, et al., Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology, Genet. Med., 17 (2015), 405–424. https://doi.org/10.1038/gim.2015.30 doi: 10.1038/gim.2015.30
|
| [18] |
S. Kundu, Mathematical model of a short translatable G-quadruplex and an assessment of its relevance to misfolding-induced proteostasis, Math. Biosci. Eng., 17 (2020), 2470–2493. https://doi.org/10.3934/mbe.2020135 doi: 10.3934/mbe.2020135
|
| [19] |
M. M. Tirion, Large amplitude elastic motions in proteins from a single-parameter, atomic analysis, Phys. Rev. Lett., 77 (1996), 1905. https://doi.org/10.1103/PhysRevLett.77.1905 doi: 10.1103/PhysRevLett.77.1905
|
| [20] |
A. R. Atilgan, S. R. Durell, R. L. Jernigan, M. C. Demirel, O. Keskin, I. Bahar, Anisotropy of fluctuation dynamics of proteins with an elastic network model, Biophys. J., 80 (2001), 505–515. https://doi.org/10.1016/S0006-3495(01)76033-X doi: 10.1016/S0006-3495(01)76033-X
|
| [21] |
P. Doruker, A. R. Atilgan, I. Bahar, Dynamics of proteins predicted by molecular dynamics simulations and analytical approaches: application to alpha-amylase inhibitor, Proteins, 40 (2000), 512–524. https://doi.org/10.1002/1097-0134(20000815)40:3<512::AID-PROT180>3.0.CO;2-M doi: 10.1002/1097-0134(20000815)40:3<512::AID-PROT180>3.0.CO;2-M
|
| [22] |
I. Bahar, A. R. Atilgan, B. Erman, Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential, Fold. Des., 2 (1997), 173–181. https://doi.org/10.1016/S1359-0278(97)00024-2 doi: 10.1016/S1359-0278(97)00024-2
|
| [23] |
K. Hinsen, Analysis of domain motions by approximate normal mode calculations, Proteins, 33 (1998), 417–429. https://doi.org/10.1002/(SICI)1097-0134(19981115)33:3<417::AID-PROT10>3.0.CO;2-8 doi: 10.1002/(SICI)1097-0134(19981115)33:3<417::AID-PROT10>3.0.CO;2-8
|
| [24] |
S. Kundu, Insights into the mechanism(s) of digestion of crystalline cellulose by plant class C GH9 endoglucanases, J. Mol. Model., 25 (2019), 240. https://doi.org/10.1007/s00894-019-4133-1 doi: 10.1007/s00894-019-4133-1
|
| [25] |
L. Yang, G. Song, R. L. Jernigan, Protein elastic network models and the ranges of cooperativity, PNAS, 106 (2009), 12347–12352. https://doi.org/10.1073/pnas.0902159106 doi: 10.1073/pnas.0902159106
|
| [26] |
X. Du, Y. Li, Y. L. Xia, S. Ai, J. Liang, P. Sang, et al., Insights into protein-ligand interactions: mechanisms, models, and methods, Int. J. Mol. Sci., 17 (2016), 144. https://doi.org/10.3390/ijms17020144 doi: 10.3390/ijms17020144
|
| [27] |
P. L. de Micheaux, B. Liquet, Understanding convergence concepts: A visual-minded and graphical simulation-based approach, Am. Stat., 63 (2009), 173–178. https://doi.org/10.1198/tas.2009.0032 doi: 10.1198/tas.2009.0032
|
| [28] |
S. Chaturvedi, A. K. Singh, A. K. Keshari, S. Maity, S. Sarkar, S. Saha, Human metabolic enzymes deficiency: A genetic mutation based approach, Scientifica (Cairo), 2016 (2016), 9828672. https://doi.org/10.1155/2016/9828672 doi: 10.1155/2016/9828672
|
| [29] |
S. Kundu, Fe(2)OG: An integrated HMM profile-based web server to predict and analyze putative non-haem iron(Ⅱ)- and 2-oxoglutarate-dependent dioxygenase function in protein sequences, BMC Res. Notes, 14 (2021), 80. https://doi.org/10.1186/s13104-021-05477-z doi: 10.1186/s13104-021-05477-z
|
| [30] |
R. J. Wanders, J. C. Komen, Peroxisomes, Refsum's disease and the alpha- and omega-oxidation of phytanic acid, Biochem. Soc. Trans., 35 (2007), 865–869. https://doi.org/10.1042/BST0350865 doi: 10.1042/BST0350865
|
| [31] |
M. A. McDonough, K. L. Kavanagh, D. Butler, T. Searls, U. Oppermann, C. J. Schofield, Structure of human phytanoyl-CoA 2-hydroxylase identifies molecular mechanisms of Refsum disease, J. Biol. Chem., 280 (2005), 41101–41110. https://doi.org/10.1074/jbc.M507528200 doi: 10.1074/jbc.M507528200
|
| [32] |
T. G. Smith, P. A. Robbins, P. J. Ratcliffe, The human side of hypoxia-inducible factor, Br. J. Haematol., 141 (2008), 325–334. https://doi.org/10.1111/j.1365-2141.2008.07029.x doi: 10.1111/j.1365-2141.2008.07029.x
|
| [33] |
G. L. Wang, G. L. Semenza, Purification and characterization of hypoxia-inducible factor 1, J. Biol. Chem., 270 (1995), 1230–1237. https://doi.org/10.1074/jbc.270.3.1230 doi: 10.1074/jbc.270.3.1230
|
| [34] |
S. E. Wilkins, M. I. Abboud, R. L. Hancock, C. J. Schofield, Targeting protein-protein interactions in the HIF system, ChemMedChem, 11 (2016), 773–786. https://doi.org/10.1002/cmdc.201600012 doi: 10.1002/cmdc.201600012
|
| [35] |
M. A. McDonough, V. Li, E. Flashman, R. Chowdhury, C. Mohr, B. M. R. Liénard, et al., Cellular oxygen sensing: Crystal structure of hypoxia-inducible factor prolyl hydroxylase (PHD2), PNAS, 103 (2006), 9814–9819. https://doi.org/10.1073/pnas.0601283103 doi: 10.1073/pnas.0601283103
|
| [36] |
P. H. Maxwell, M. S. Wiesener, G. W. Chang, S. C. Clifford, E. C. Vaux, M. E. Cockman, et al., The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis, Nature, 399 (1999), 271–275. https://doi.org/10.1038/20459 doi: 10.1038/20459
|
| [37] |
G. L. Semenza, Hydroxylation of HIF-1: oxygen sensing at the molecular level, Physiology (Bethesda), 19 (2004), 176–182. https://doi.org/10.1152/physiol.00001.2004 doi: 10.1152/physiol.00001.2004
|
| [38] |
D. R. Peaper, P. Cresswell, Regulation of MHC class Ⅰ assembly and peptide binding, Annu. Rev. Cell Dev. Biol., 24 (2008), 343–368. https://doi.org/10.1146/annurev.cellbio.24.110707.175347 doi: 10.1146/annurev.cellbio.24.110707.175347
|
| [39] |
E. W. Hewitt, The MHC class Ⅰ antigen presentation pathway: strategies for viral immune evasion, Immunology, 110 (2003), 163–169. https://doi.org/10.1046/j.1365-2567.2003.01738.x doi: 10.1046/j.1365-2567.2003.01738.x
|
| [40] |
E. Rufer, R. M. Leonhardt, M. R. Knittler, Molecular architecture of the TAP-associated MHC class Ⅰ peptide-loading complex, J. Immunol., 179 (2007), 5717–5727. https://doi.org/10.4049/jimmunol.179.9.5717 doi: 10.4049/jimmunol.179.9.5717
|
| [41] |
A. Blees, D. Januliene, T. Hofmann, N. Koller, C. Schmidt, S. Trowitzsch, et al., Structure of the human MHC-Ⅰ peptide-loading complex, Nature, 551 (2017), 525–528. https://doi.org/10.1038/nature24627 doi: 10.1038/nature24627
|
| [42] |
J. W. Yewdell, J. R. Bennink, Immunodominance in major histocompatibility complex class Ⅰ-restricted T lymphocyte responses, Annu. Rev. Immunol., 17 (1999), 51–88. https://doi.org/10.1146/annurev.immunol.17.1.51 doi: 10.1146/annurev.immunol.17.1.51
|
| [43] |
P. V. Praveen, R. Yaneva, H. Kalbacher, S. Springer, Tapasin edits peptides on MHC class Ⅰ molecules by accelerating peptide exchange, Eur. J. Immunol., 40 (2010), 214–224. https://doi.org/10.1002/eji.200939342 doi: 10.1002/eji.200939342
|
| [44] |
S. Kundu, Mathematical modeling and stochastic simulations suggest that low-affinity peptides can bisect MHC1-mediated export of high-affinity peptides into "early"- and "late"-phases, Heliyon, 7 (2021), e07466. https://doi.org/10.1016/j.heliyon.2021.e07466 doi: 10.1016/j.heliyon.2021.e07466
|
 mbe-19-12-620-S01.rar mbe-19-12-620-S01.rar |
 |
Figures(1) / Tables(2)

Siddhartha Kundu. Modeling ligand-macromolecular interactions as eigenvalue-based transition-state dissociation constants may offer insights into biochemical function of the resulting complexes[J]. Mathematical Biosciences and Engineering, 2022, 19(12): 13252-13275. doi: 10.3934/mbe.2022620







 DownLoad:
DownLoad: