Citation: Linlu Song, Shangbo Ning, Jinxuan Hou, Yunjie Zhao. Performance of protein-ligand docking with CDK4/6 inhibitors: a case study[J]. Mathematical Biosciences and Engineering, 2021, 18(1): 456-470. doi: 10.3934/mbe.2021025

| [1] | S. Pernas, S. M. Tolaney, E. P. Winer, S. Goel, CDK4/6 inhibition in breast cancer: current practice and future directions, Ther. Adv. Med. Oncol., 10 (2018), 1758835918786451. |
| [2] |
H. Xu, S. Yu, Q. Liu, X. Yuan, S. Mani, R. G. Pestell, et al., Recent advances of highly selective CDK4/6 inhibitors in breast cancer, J. Hematol. Oncol., 10 (2017), 97. doi: 10.1186/s13045-017-0467-2

|
| [3] | S. F. Dowdy, M. Kaulich, Abstract 1304: Cyclin D:Cdk4/6 activates RB by mono-phosphorylation during early G1 phase, Cancer Res., 74 (2014), 1304–1304. |
| [4] | A. N. Omstead, D. Matsui, J. E. Kosovec, S. A. Martin, B. A. Jobe, Antitumor efficacy of CDK 4/6 dual inhibitor, abemaciclib, in an esophageal adenocarcinoma model, J. Clin. Oncol., 35 (2017), e15598–e15598. |
| [5] |
M. Tutone, A. M. Almerico, Recent advances on CDK inhibitors: An insight by means of in silico methods, Eur. J. Med. Chem., 142 (2017), 300–315. doi: 10.1016/j.ejmech.2017.07.067
|
| [6] |
S. Müller, A. Chaikuad, N. S. Gray, S. Knapp, The ins and outs of selective kinase inhibitor development, Nat. Chem. Biol., 11 (2015), 818–821. doi: 10.1038/nchembio.1938
|
| [7] | P. Ayaz, D. Andres, D. A. Kwiatkowski, C. C. Kolbe, P. Lienau, G. Siemeister, et al., Conformational adaption may explain the slow dissociation kinetics of roniciclib (BAY 1000394), a yype I CDK inhibitor with kinetic selectivity for CDK2 and CDK9, ACS Chem. Biol., (2016), acschembio.6b00074. |
| [8] | T. Dale, P. A. Clarke, C. Esdar, D. Waalboer, O. Adeniji-Popoola, M. J. Ortiz-Ruiz, et al., A selective chemical probe for exploring the role of CDK8 and CDK19 in human disease, Nat. Chem. Biol., 2015. |
| [9] | M. Schreuer, V. Kruse, Y. Jansen, B. Neyns, COMBI-rechallenge: a phase II clinical trial on dabrafenib plus trametinib in BRAFV600-mutant melanoma patients who previously experienced progression on BRAF(+MEK)-inhibition, Ann. Oncolo., 27 (2016). |
| [10] | R. B. Corcoran, G. S. Falchook, J. R. Infante, O. Hamid, W. A. Messersmith, E. L. Kwak, et al., BRAF V600 mutant colorectal cancer (CRC) expansion cohort from the phase I/II clinical trial of BRAF inhibitor dabrafenib (GSK2118436) plus MEK inhibitor trametinib (GSK1120212), J. Clin. Oncol., 2012. |
| [11] |
T. Wang, Z. Yang, Y. Zhang, W. Yan, F. Wang, L. He, et al., Discovery of novel CDK8 inhibitors using multiple crystal structures in docking-based virtual screening, Eur. J. Med. Chem., 129 (2017), 275–286. doi: 10.1016/j.ejmech.2017.02.020
|
| [12] |
S. E. Dixon-Clarke, S. N. Shehata, T. Krojer, T. D. Sharpe, F. Von Delft, K. Sakamoto, et al., Structure and inhibitor specificity of the PCTAIRE-family kinase CDK16, Biochem. J., 474 (2017), 699–713. doi: 10.1042/BCJ20160941
|
| [13] |
N. Canela, M. Orzaez, R. Fucho, F. Mateo, R. Gutierrez, A. Pineda-Lucena, et al., Identification of an hexapeptide that binds to a surface pocket in cyclin A and inhibits the catalytic activity of the complex cyclin-dependent kinase 2-cyclin A, J. Biol. Chem., 281 (2006), 35942–35953. doi: 10.1074/jbc.M603511200
|
| [14] | Orzáez, Guevara, Sancho, Pérez-Payá, Intrinsic caspase-8 activation mediates sensitization of erlotinib-resistant tumor cells to erlotinib/cell-cycle inhibitors combination treatment, Cell Death Dis., 2012. |
| [15] |
R. S. Finn, A. Aleshin, D. J. Slamon, Targeting the cyclin-dependent kinases (CDK) 4/6 in estrogen receptor-positive breast cancers, Breast Cancer Res.: BCR, 18 (2016), 17. doi: 10.1186/s13058-015-0661-5
|
| [16] |
T. Otto, P. Sicinski, Cell cycle proteins as promising targets in cancer therapy, Nat. Rev. Cancer, 17 (2017), 93–115. doi: 10.1038/nrc.2016.138
|
| [17] | L. Spring, A. Bardia, S. Modi, Targeting the cyclin D-cyclin-dependent kinase (CDK) 4/6-retinoblastoma pathway with selective CDK 4/6 inhibitors in hormone receptor-positive breast cancer: rationale, current status, and future directions, Discovery Med., 21 (2016), 65. |
| [18] |
M. W. Landis, B. S. Pawlyk, T. Li, P. Sicinski, P. W. Hinds, Cyclin D1-dependent kinase activity in murine development and mammary tumorigenesis, Cancer Cell, 9 (2006), 13–22. doi: 10.1016/j.ccr.2005.12.019
|
| [19] | B. Laderian, T. Fojo, CDK4/6 inhibition as a therapeutic strategy in breast cancer: palbociclib, ribociclib, and abemaciclib, Semin. Oncol., (2018), S0093775418300812. |
| [20] | S. Parylo, A. Vennepureddy, V. Dhar, P. Patibandla, A. Sokoloff, Role of cyclin-dependent kinase 4/6 inhibitors in the current and future eras of cancer treatment, J. Oncol. Pharm. Pract., (2018), 107815521877090. |
| [21] | A. Patnaik, L. S. Rosen, S. M. Tolaney, A. W. Tolcher, J. W. Goldman, L. Gandhi, et al., Efficacy and safety of abemaciclib, an inhibitor of CDK4 and CDK6, for patients with breast cancer, non–small cell lung cancer, and other solid tumors, Cancer Discovery, (2016), 740–753. |
| [22] | H. Wang, K. Wang, Z. Guan, Y. Jian, Y. Jia, F. Kashanchi, et al., Computational study of non-catalytic T-loop pocket on CDK proteins for drug development, Chin. Phys. B, 2017. |
| [23] | H. W. Wang, Z. Y. Guan, J. D. Qiu, Y. Jia, C. Zeng, Y. J. Zhao, Novel method to identify group-specific non-catalytic pockets of human kinome for drug design, RSC Adv., 4 (2020). |
| [24] | Y. Zhao, H. Chen, C. Du, Y. Jian, H. Li, Y. Xiao, et al., Design of tat-activated CDK9 inhibitor, Int. J. Peptide Res. Therapeutics, 25 (2018), 807–817. |
| [25] |
A. M. Almerico, M. Tutone, A. Lauria, 3D-QSAR pharmacophore modeling and in silico screening of new Bcl-xl inhibitors, Eur. J. Med. Chem., 45 (2010), 4774–4782. doi: 10.1016/j.ejmech.2010.07.042
|
| [26] |
A. M. Almerico, M. Tutone, A. Lauria, Receptor-guided 3D-QSAR approach for the discovery of c-kit tyrosine kinase inhibitors, J. Mol. Model., 18 (2012), 2885–2895. doi: 10.1007/s00894-011-1304-0
|
| [27] | Z. Shentu, M. A. Hasan, C. Bystroff, M. J. Zaki, Context shapes: Efficient complementary shape matching for protein-protein docking, Proteins-Struct. Funct. Bioinformatics, 70 (2010), 1056–1073. |
| [28] | D. W. Ritchie, Evaluation of protein docking predictions using Hex 3.1 in CAPRI rounds 1 and 2, Proteins: Struct., Funct., Bioinformatics, 2003. |
| [29] |
K. Wiehe, B. Pierce, J. Mintseris, W. W. Tong, R. Anderson, R. Chen, et al., ZDOCK and RDOCK performance in CAPRI rounds 3, 4, and 5, Proteins-Struct. Funct. Bioinformatics, 60 (2005), 207–213. doi: 10.1002/prot.20559
|
| [30] | A. Caflisch, P. Niederer, M. Anliker, Monte Carlo docking of oligopeptides to proteins, Proteins-Struct. Funct. Bioinformatics, 13 (2010), 223–230. |
| [31] | T. N. Hart, R. J. Read, A multiple-start Monte Carlo docking method, J. Mol. Graphics, 13 (2010), 206–222. |
| [32] | P. Reigan, W. Guo, D. Siegel, D. Ross, Molecular docking studies investigating the interaction of a series of benzoquinone ansamycin Hsp90 inhibitors with NAD(P)H: quinone oxidoreductase 1 (NQO1), Cancer Res., 66 (2006), 457–457. |
| [33] |
C. M. Venkatachalam, X. Jiang, T. Oldfield, M. Waldman, LigandFit: a novel method for the shape-directed rapid docking of ligands to protein active sites, J. Mol. Graphics Model., 21 (2003), 289–307. doi: 10.1016/S1093-3263(02)00164-X
|
| [34] | L. Kang, H. L. Li, H. L. Jiang, X. C. Wang, An improved adaptive genetic algorithm for protein–ligand docking, J. Comput. Aided Mol. Des., 23 (2009), 1–12. |
| [35] |
F. Sterberg, G. M. Morris, M. F. Sanner, A. J. Olson, D. S. Goodsell, Automated docking to multiple target structures: Incorporation of protein mobility and structural water heterogeneity in AutoDock, Protns Struct. Funct. Bioinformatics, 46 (2002), 34–40. doi: 10.1002/prot.10028
|
| [36] |
G. Jones, P. Willett, R. C. Glen, A. R. Leach, R. Taylor, Development and validation of a genetic algorithm for flexible docking, J. Mol. Biol., 267 (1997), 727–748. doi: 10.1006/jmbi.1996.0897
|
| [37] |
H. Jing, X. Zhou, X. Dong, J. Cao, H. Zhu, J. Lou, et al., Abrogation of Akt signaling by Isobavachalcone contributes to its anti-proliferative effects towards human cancer cells, Cancer Lett., 294 (2010), 167–177. doi: 10.1016/j.canlet.2010.01.035
|
| [38] |
H. Li, C. Li, C. Gui, X. Luo, K. Chen, J. Shen, et al., GAsDock: a new approach for rapid flexible docking based on an improved multi-population genetic algorithm, Bioorg. Med. Chem. Lett., 14 (2004), 4671–4676. doi: 10.1016/j.bmcl.2004.06.091
|
| [39] | G. Culletta, A. M. Almerico, M. Tutone, Comparing molecular dynamics-derived pharmacophore models with docking: a study on CDK-2 inhibitors, Chem. Data Collect., 2020. |
| [40] | P. N. Sekhar, Software for molecular docking: a review, Biophys. Rev., 9 (2016), 91–102. |
| [41] |
Z. Bikadi, E. Hazai, Application of the PM6 semi-empirical method to modeling proteins enhances docking accuracy of AutoDock, J. Cheminformatics, 1 (2009), 1–16. doi: 10.1186/1758-2946-1-1
|
| [42] | A. Grosdidier, V. Zoete, O. Michielin, SwissDock, a protein-small molecule docking web service based on EADock DSS, Nucleic Acids Res., 39 (2011), W270–W277. |
| [43] | G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, A. J. Olson, AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility, J. Comput. Chem., 30 (2010), 2785–2791. |
| [44] |
A. Grosdidier, V. Zoete, O. Michielin, Fast docking using the CHARMM force field with EADock DSS, J. Comput. Chem., 32 (2011), 2149–2159. doi: 10.1002/jcc.21797
|
| [45] | R. Huey, G. M. Morris, A. J. Olson, D. S. Goodsell, A semi-empirical free energy force field with charge-based desolvation, J. Comput. Chem., 28 (2010), 1145–1152. |
| [46] |
S. J. Weiner, P. A. Kollman, D. A. Case, U. C. Singh, C. Ghio, G. Alagona, et al., A new force field for molecular mechanical simulation of nucleic acids and proteins, J. Am. Chem. Soc., 106 (1984), 765–784. doi: 10.1021/ja00315a051
|
| [47] |
P. J. Goodford, A computational procedure for determining energetically favorable binding sites on biologically important macromolecules, J. Med. Chem., 28 (1985), 849–857. doi: 10.1021/jm00145a002
|
| [48] | E. L. Mehler, T. Solmajer, Electrostatic effects in proteins: comparison of dielectric and charge models, Protn. Eng., (1991), 903–910. |
| [49] |
G. M. Verkhivker, D. Bouzida, D. K. Gehlhaar, P. A. Rejto, S. Arthurs, A. B. Colson, et al., Deciphering common failures in molecular docking of ligand-protein complexes, J. Comput.-Aided Mol. Des., 14 (2000), 731–751. doi: 10.1023/A:1008158231558
|
| [50] | B. R. Brooks, R. E. Bruccoleri, B. D. Olafson, D. J. States, M. Karplus, CHARMM: A program for macromolecular energy, minimization, and dynamics calculations, J. Comput. Chem., 4 (2010), 187–217. |
| [51] |
A. Grosdidier, V. Zoete, O. Michielin, Fast docking using the CHARMM force field with EADock DSS, J. Comput. Chem., 32 (2011), 2149–2159. doi: 10.1002/jcc.21797
|
| [52] |
H. J. C. Berendsen, J. R. Grigera, T. P. Straatsma, The missing term in effective pair potentials, J. Phys. Chem., 91 (1987), 6269–6271. doi: 10.1021/j100308a038
|
| [53] |
B. R. R. Brooks, C. L. B. Brooks, A. D. Mackerell, L. Nilsson, M. J. Karplus, CHARMM: the biomolecular simulation program, J. Comput. Chem., 30 (2009), 1545. doi: 10.1002/jcc.21287
|
| [54] | J. A. Hartigan, M. A. Wong, A K-Means clustering algorithm, Appl. Stats., 28 (1979). |
| [55] |
A. K. Jain, Data clustering: 50 years beyond K-means, Pattern Recognit. Lett., 31 (2010), 651–666. doi: 10.1016/j.patrec.2009.09.011
|
| [56] | A. B. Chorin, G. Masrati, A. Kessel, A. Narunsky, ConSurf‐DB: An accessible repository for the evolutionary conservation patterns of the majority of PDB proteins, Protein Sci., 29 (2020). |
| [57] |
O. Goldenberg, E. Erez, G. Nimrod, N. Ben-Tal, The ConSurf-DB: pre-calculated evolutionary conservation profiles of protein structures, Nucleic Acids Res., 37 (2009), D323–D327. doi: 10.1093/nar/gkn822
|
| [58] |
M. Jaina, R. D. Finn, S. R. Eddy, B. Alex, P. Marco, Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions, Nucleic Acids Res., 41 (2013), e121–e121. doi: 10.1093/nar/gkt263
|
| [59] |
K. Kazutaka, D. M. Standley, MAFFT multiple sequence alignment software version 7: improvements in performance and usability, Mol. Biol. Evol., 30 (2013), 772–780. doi: 10.1093/molbev/mst010
|
| [60] | P. Tal, R. E. Bell, M. Itay, G. Fabian, B. T. Nir, Rate4Site: an algorithmic tool for the identification of functional regions in proteins by surface mapping of evolutionary determinants within their homologues, Bioinformatics, (2002), S71. |
| [61] |
P. Chen, N. V. Lee, W. Hu, M. Xu, B. W. Murray, Spectrum and degree of CDK drug interactions predicts clinical performance, Mol. Cancer Therapeutics, 15 (2016), 2273. doi: 10.1158/1535-7163.MCT-16-0300
|
| [62] |
N. M. O'Boyle, M. Banck, C. A. James, C. Morley, G. R. Hutchison, Open babel: an open chemical toolbox, J. Cheminformatics, 3 (2011), 33. doi: 10.1186/1758-2946-3-33
|
| [63] |
H. Wang, J. Qiu, H. Liu, Y. Xu, Y. Jia, Y. Zhao, HKPocket: human kinase pocket database for drug design, BMC Bioinformatics, 20 (2019), 617. doi: 10.1186/s12859-019-3254-y
|
| [64] | K. Wang, Y. Jian, H. Wang, C. Zeng, Y. Zhao, RBind: computational network method to predict RNA binding sites, Bioinformatics, 34 (2018). |
| [65] |
Y. Jian, X. Wang, J. Qiu, H. Wang, Z. Liu, Y. Zhao, C. Zeng, DIRECT: RNA contact predictions by integrating structural patterns, BMC Bioinformatics, 20 (2019), 497. doi: 10.1186/s12859-019-3099-4
|
| [66] |
H. Wang, Y. Zhao, RBinds: a user-friendly server for RNA binding site prediction, Comput. Struct. Biotechnol. J., 18 (2020), 3762–3765. doi: 10.1016/j.csbj.2020.10.043
|
| [67] |
H. Wang, Y. Zhao, Methods and applications of RNA contact prediction, Chin. Phys. B, 29 (2020), 108708. doi: 10.1088/1674-1056/abb7f3
|
| [68] | K. Rascon, G. Flajc, C. De Angelis, X. Liu, M. V. Trivedi, E. Ekinci, Ribociclib in HR+/HER2- advanced or metastatic breast cancer patients, Ann. Pharmacotherapy, 2019. |
| [69] | R. J. Cersosimo, Cyclin-dependent kinase 4/6 inhibitors for the management of advanced or metastatic breast cancer in women, (2019), 1183–1202. |
| [70] |
A. F. D. Groot, C. J. Kuijpers, J. R. Kroep, CDK4/6 inhibition in early and metastatic breast cancer: A review, Cancer Treatment Rev., 60 (2017), 130–138. doi: 10.1016/j.ctrv.2017.09.003
|
| [71] |
M. Poratti, G. Marzaro, Third-generation CDK inhibitors: A review on the synthesis and binding modes of Palbociclib, Ribociclib and Abemaciclib, Eur. J. Med. Chem., 172 (2019), 143–153. doi: 10.1016/j.ejmech.2019.03.064
|
 mbe-18-01-025-supplementary.pdf mbe-18-01-025-supplementary.pdf |
 |
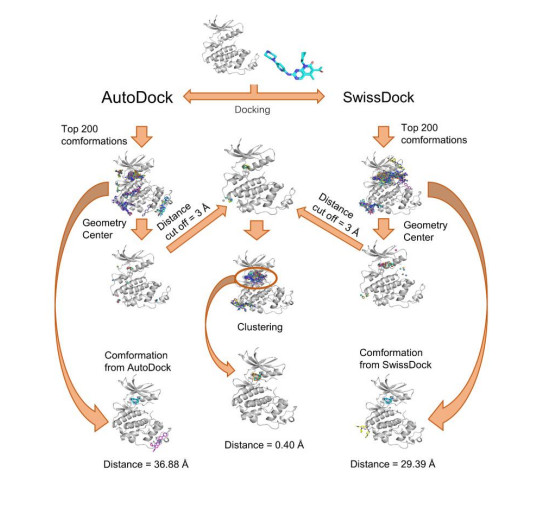
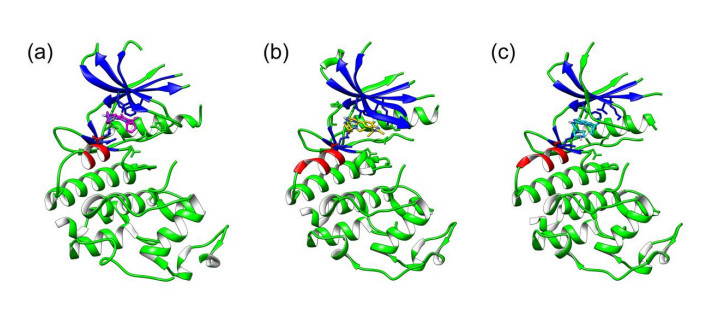
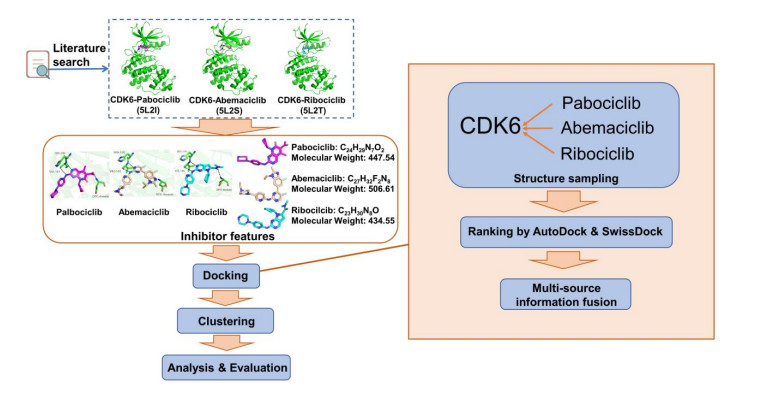
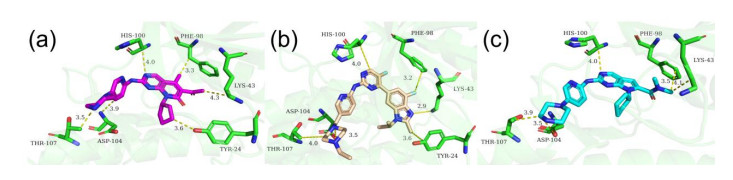
Figures(7)

Linlu Song, Shangbo Ning, Jinxuan Hou, Yunjie Zhao. Performance of protein-ligand docking with CDK4/6 inhibitors: a case study[J]. Mathematical Biosciences and Engineering, 2021, 18(1): 456-470. doi: 10.3934/mbe.2021025







 DownLoad:
DownLoad: