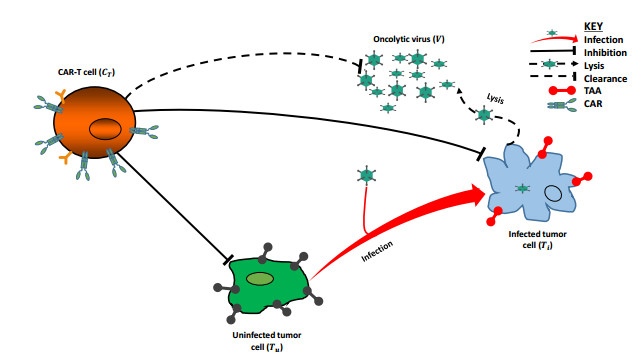
Combining chimeric antigen receptor T (CAR-T) cells with oncolytic viruses (OVs) has recently emerged as a promising treatment approach in preclinical studies that aim to alleviate some of the barriers faced by CAR-T cell therapy. In this study, we address by means of mathematical modeling the main question of whether a single dose or multiple sequential doses of CAR-T cells during the OVs therapy can have a synergetic effect on tumor reduction. To that end, we propose an ordinary differential equations-based model with virus-induced synergism to investigate potential effects of different regimes that could result in efficacious combination therapy against tumor cell populations. Model simulations show that, while the treatment with a single dose of CAR-T cells is inadequate to eliminate all tumor cells, combining the same dose with a single dose of OVs can successfully eliminate the tumor in the absence of virus-induced synergism. However, in the presence of virus-induced synergism, the same combination therapy fails to eliminate the tumor. Furthermore, it is shown that if the intensity of virus-induced synergy and/or virus oncolytic potency is high, then the induced CAR-T cell response can inhibit virus oncolysis. Additionally, the simulations show a more robust synergistic effect on tumor cell reduction when OVs and CAR-T cells are administered simultaneously compared to the combination treatment where CAR-T cells are administered first or after OV injection. Our findings suggest that the combination therapy of CAR-T cells and OVs seems unlikely to be effective if the virus-induced synergistic effects are included when genetically engineering oncolytic viral vectors.
Citation: Khaphetsi Joseph Mahasa, Rachid Ouifki, Amina Eladdadi, Lisette de Pillis. A combination therapy of oncolytic viruses and chimeric antigen receptor T cells: a mathematical model proof-of-concept[J]. Mathematical Biosciences and Engineering, 2022, 19(5): 4429-4457. doi: 10.3934/mbe.2022205
Combining chimeric antigen receptor T (CAR-T) cells with oncolytic viruses (OVs) has recently emerged as a promising treatment approach in preclinical studies that aim to alleviate some of the barriers faced by CAR-T cell therapy. In this study, we address by means of mathematical modeling the main question of whether a single dose or multiple sequential doses of CAR-T cells during the OVs therapy can have a synergetic effect on tumor reduction. To that end, we propose an ordinary differential equations-based model with virus-induced synergism to investigate potential effects of different regimes that could result in efficacious combination therapy against tumor cell populations. Model simulations show that, while the treatment with a single dose of CAR-T cells is inadequate to eliminate all tumor cells, combining the same dose with a single dose of OVs can successfully eliminate the tumor in the absence of virus-induced synergism. However, in the presence of virus-induced synergism, the same combination therapy fails to eliminate the tumor. Furthermore, it is shown that if the intensity of virus-induced synergy and/or virus oncolytic potency is high, then the induced CAR-T cell response can inhibit virus oncolysis. Additionally, the simulations show a more robust synergistic effect on tumor cell reduction when OVs and CAR-T cells are administered simultaneously compared to the combination treatment where CAR-T cells are administered first or after OV injection. Our findings suggest that the combination therapy of CAR-T cells and OVs seems unlikely to be effective if the virus-induced synergistic effects are included when genetically engineering oncolytic viral vectors.

| [1] |
N. Nishio, I. Diaconu, H. Liu, V. Cerullo, I. Caruana, V. Hoyos, et al., Armed oncolytic virus enhances immune functions of chimeric antigen receptormodified T cells in solid tumors, Cancer Res., 74 (2014), 5195–5205. https://doi.org/10.1158/0008-5472.CAN-14-0697 doi: 10.1158/0008-5472.CAN-14-0697

|
| [2] |
K. Tanoue, A. R. Shaw, N. Watanabe, C. Porter, B. Rana, S. Gottschalk, et al., Armed oncolytic adenovirus-expressing PD-L1 mini-body enhances antitumor effects of chimeric antigen receptor T cells in solid tumors, Cancer Res., 77 (2017), 2040–2051. https://doi.org/10.1158/0008-5472.CAN-16-1577 doi: 10.1158/0008-5472.CAN-16-1577
|
| [3] |
A. R. Shaw, C. E. Porter, N. Watanabe, K. Tanoue, A. Sikora, S. Gottschalk, et al., Adenovirotherapy delivering cytokine and checkpoint inhibitor augments CAR T cells against metastatic head and neck cancer, Mol. Ther., 25 (2017), 2440–2451. https://doi.org/10.1016/j.ymthe.2017.09.010 doi: 10.1016/j.ymthe.2017.09.010
|
| [4] |
S. J. Schuster, M. R. Bishop, C. S. Tam, E. K. Waller, P. Borchmann, J. P. McGuirk, et al., Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma., N. Engl. J. Med., 380 (2019), 45–56. https://doi.org/10.1007/s00134-018-05509-6 doi: 10.1007/s00134-018-05509-6
|
| [5] |
R. Huang, X. Li, Y. He, W. Zhu, L. Gao, Y. Liu, et al., Recent advances in CAR-T cell engineering, J. Hematol. Oncol., 13 (2020), 1–9. https://doi.org/10.1186/s13045-019-0838-y doi: 10.1186/s13045-019-0838-y
|
| [6] |
E. Jacoby, Relapse and resistance to CAR-T cells and blinatumomab in hematologic malignancies, Clin. Hematol. Int., 1 (2019), 79–84. https://doi.org/10.2991/chi.d.190219.001 doi: 10.2991/chi.d.190219.001
|
| [7] |
N. Albinger, J. Hartmann, E. Ullrich, Current status and perspective of CAR-T and CAR-NK cell therapy trials in Germany, Gene Ther., (2021), 1–15. https://doi.org/10.1038/s41434-021-00246-w doi: 10.1038/s41434-021-00246-w
|
| [8] |
Y. Guo, K. Feng, Y. Wang, W. Han, Targeting cancer stem cells by using chimeric antigen receptor-modified T cells: a potential and curable approach for cancer treatment, Protein cell, 9 (2018), 516–526. https://doi.org/10.1007/s13238-017-0394-6 doi: 10.1007/s13238-017-0394-6
|
| [9] |
A. K. Park, Y. Fong, S. I. Kim, J. Yang, J. P. Murad, J. Lu, et al., Effective combination immunotherapy using oncolytic viruses to deliver CAR targets to solid tumors, Sci. Transl. Med., 12 (2020), eaaz1863. https://doi.org/10.1126/scitranslmed.aaz1863 doi: 10.1126/scitranslmed.aaz1863
|
| [10] |
D. N. Khalil, E. L. Smith, R. J. Brentjens, J. D. Wolchok, The future of cancer treatment: immunomodulation, CARs and combination immunotherapy, Nat. Rev. Clin. Oncol., 13 (2016), 273–290. https://doi.org/10.1038/nrclinonc.2016.25 doi: 10.1038/nrclinonc.2016.25
|
| [11] |
A. Wing, C. A. Fajardo, A. D. J. Posey, C. Shaw, T. Da, R. M. Young, et al., Improving CART-cell therapy of solid tumors with oncolytic virus-driven production of a bispecific T-cell engager, Cancer Immunol. Res., 6 (2018), 605–616. https://doi.org/10.1158/2326-6066.CIR-17-0314 doi: 10.1158/2326-6066.CIR-17-0314
|
| [12] |
G. V. Kochneva, G. F. Sivolobova, A. V. Tkacheva, A. A. Gorchakov, S. V. Kulemzin, Combination of oncolytic virotherapy and CAR T/NK cell therapy for the treatment of cancer, Mol. Biol., 54 (2020), 1–12. https://doi.org/10.1134/S0026893320010100 doi: 10.1134/S0026893320010100
|
| [13] |
L. Zhao, Y. J. Cao, Engineered T cell therapy for cancer in the clinic, Front. Immunol., 10 (2019), 2250. https://doi.org/10.3389/fimmu.2019.02250 doi: 10.3389/fimmu.2019.02250
|
| [14] |
K. J. Mahasa, R. Ouifki, A. Eladdadi, L. de Pillis, Mathematical model of tumor-immune surveillance, J. Theor. Biol., 404 (2016), 312–330. https://doi.org/10.1016/j.jtbi.2016.06.012 doi: 10.1016/j.jtbi.2016.06.012
|
| [15] |
R. G. Majzner, C. L. Mackall, Tumor antigen escape from CAR T-cell therapy, Cancer Discovery, 8 (2018), 1219–1226. https://doi.org/10.1158/2159-8290.CD-18-0442 doi: 10.1158/2159-8290.CD-18-0442
|
| [16] |
R. C. Sterner, R. M. Sterner, CAR-T cell therapy: current limitations and potential strategies, Blood Cancer J., 11 (2021), 1–11. https://doi.org/10.1038/s41408-021-00459-7 doi: 10.1038/s41408-021-00459-7
|
| [17] |
A. R. Yoon, J. Hong, Y. Li, H. C. Shin, H. Lee, H. S. Kim, et al., Mesenchymal stem cell-mediated delivery of an oncolytic adenovirus enhances antitumor efficacy in hepatocellular carcinoma, Cancer Res., 79 (2019), 4503–4514. https://doi.org/10.1158/0008-5472.CAN-18-3900 doi: 10.1158/0008-5472.CAN-18-3900
|
| [18] |
K. J. Mahasa, L. de Pillis, R. Ouifki, A. Eladdadi, P. Maini, A. R. Yoon, et al., Mesenchymal stem cells used as carrier cells of oncolytic adenovirus results in enhanced oncolytic virotherapy, Sci. Rep., 10 (2020), 1–13. https://doi.org/10.1038/s41598-019-56847-4 doi: 10.1038/s41598-019-56847-4
|
| [19] |
J. Gao, W. Zhang, A. Ehrhardt, Expanding the spectrum of adenoviral vectors for cancer therapy, Cancers, 12 (2020), 1139. https://doi.org/10.3390/cancers12051139 doi: 10.3390/cancers12051139
|
| [20] |
L. Russell, K. W. Peng, S. J. Russell, R. M. Diaz, Oncolytic viruses: priming time for cancer immunotherapy, BioDrugs, 33 (2019), 485–501. https://doi.org/10.1007/s40259-019-00367-0 doi: 10.1007/s40259-019-00367-0
|
| [21] |
H. L. Kaufman, F. J. Kohlhapp, A. Zloza, Oncolytic viruses: a new class of immunotherapy drugs, Nat. Rev. Drug Discovery, 14 (2015), 642–662. https://doi.org/10.1038/nrd4663 doi: 10.1038/nrd4663
|
| [22] | B. Zhang, P. Cheng, Improving antitumor efficacy via combinatorial regimens of oncolytic virotherapy, Mol. Cancer, 19 (2020), 158. |
| [23] |
Q. Zhang, F. Liu, Advances and potential pitfalls of oncolytic viruses expressing immunomodulatory transgene therapy for malignant gliomas, Cell Death Dis., 11 (2020), 1–11. https://doi.org/10.1038/s41419-020-2696-5 doi: 10.1038/s41419-020-2696-5
|
| [24] |
S. E. Lawler, M. C. Speranza, C. F. Cho, E. A. Chiocca, Oncolytic viruses in cancer treatment: a review, JAMA oncol., 3 (2017), 841–849. https://doi.org/10.1001/jamaoncol.2016.2064 doi: 10.1001/jamaoncol.2016.2064
|
| [25] |
A. H. Choi, M. P. O'Leary, Y. Fong, N. G. Chen, From benchtop to bedside: a review of oncolytic virotherapy, Biomed., 4 (2016), 18. https://doi.org/10.3390/biomedicines4030018 doi: 10.3390/biomedicines4030018
|
| [26] |
L. Aurelian, Oncolytic viruses as immunotherapy: progress and remaining challenges, OncoTargets Ther., 9 (2016), 2627–2637. https://doi.org/10.2147/OTT.S63049 doi: 10.2147/OTT.S63049
|
| [27] |
S. Zuo, M. Wei, B. He, A. Chen, S. Wang, L. Kong, et al., Enhanced antitumor efficacy of a novel oncolytic vaccinia virus encoding a fully monoclonal antibody against T-cell immunoglobulin and ITIM domain (TIGIT), EBioMedicine, 64 (2021), 103240. https://doi.org/10.1016/j.ebiom.2021.103240 doi: 10.1016/j.ebiom.2021.103240
|
| [28] |
N. Nishio, G. Dotti, Oncolytic virus expressing RANTES and IL-15 enhances function of CAR-modified T cells in solid tumors, Oncoimmunology, 4 (2015), e988098. https://doi.org/10.4161/21505594.2014.988098 doi: 10.4161/21505594.2014.988098
|
| [29] |
E. K. Moon, L. C. S. Wang, K. Bekdache, R. C. Lynn, A. Lo, S. H. Thorne, et al., Intratumoral delivery of CXCL11 via a vaccinia virus, but not by modified T cells, enhances the efficacy of adoptive T cell therapy and vaccines, Oncoimmunology, 7 (2018), e1395997. https://doi.org/10.1080/2162402X.2017.1395997 doi: 10.1080/2162402X.2017.1395997
|
| [30] |
N. Nishio, I. Diaconu, H. Liu, V. Cerullo, I. Caruana, V. Hoyos, et al., Armed oncolytic virus enhances immune functions of chimeric antigen receptor–modified T cells in solid tumors, Cancer Res., 74 (2014), 5195–5205. https://doi.org/10.1158/0008-5472.CAN-14-0697 doi: 10.1158/0008-5472.CAN-14-0697
|
| [31] | A. Ajina, J. Maher, Prospects for combined use of oncolytic viruses and CAR T cells, J. Immunother. Cancer, 5 (2017), 1–12. |
| [32] |
L. Evgin, A. L. Huff, P. Wongthida, J. Thompson, T. Kottke, J. Tonne, et al., Oncolytic virus-derived type I interferon restricts CAR T cell therapy, Nat. Commun., 11 (2020), 1–15. https://doi.org/10.1038/s41467-020-17011-z doi: 10.1038/s41467-020-17011-z
|
| [33] | S. Guedan, R. Alemany, CAR-T cells and oncolytic viruses: joining forces to overcome the solid tumor challenge, Front. Immunol., 9 (2018), 2460. |
| [34] |
L. Evgin, R. G. Vile, Parking CAR T cells in tumours: oncolytic viruses as valets or vandals, Cancers, 13 (2021), 1106. https://doi.org/10.3390/cancers13051106 doi: 10.3390/cancers13051106
|
| [35] |
A. Aalipour, F. L. Boeuf, M. Tang, S. Murty, F. Simonetta, A. X. Lozano, et al., Viral delivery of CAR targets to solid tumors enables effective cell therapy, Mol. Ther. Oncolytics, 17 (2020), 232–240. https://doi.org/10.1016/j.omto.2020.03.018 doi: 10.1016/j.omto.2020.03.018
|
| [36] |
L. Evgin, R. G. Vile, Parking CAR T cells in tumours: oncolytic viruses as valets or vandals, Cancers, 13 (2021), 1106. https://doi.org/10.3390/cancers13051106 doi: 10.3390/cancers13051106
|
| [37] |
N. Watanabe, M. K. McKenna, A. R. Shaw, M. Suzuki, Clinical CAR-T cell and oncolytic virotherapy for cancer treatment, Mol. The., 29 (2021), 505–520. https://doi.org/10.1016/j.ymthe.2020.10.023 doi: 10.1016/j.ymthe.2020.10.023
|
| [38] |
T. Shi, X. Song, Y. Wang, F. Liu, J. Wei, Combining oncolytic viruses with cancer immunotherapy: establishing a New Generation of Cancer Treatment, Front. Immunol., 11 (2020), 683. https://doi.org/10.3389/fimmu.2020.00683 doi: 10.3389/fimmu.2020.00683
|
| [39] |
X. Y. Tang, Y. S. Ding, T. Zhou, X. Wang, Y. Yang, Tumor-tagging by oncolytic viruses: A novel strategy for CAR-T therapy against solid tumors, Cancer Lett., 503 (2021), 69–74. https://doi.org/10.1016/j.canlet.2021.01.014 doi: 10.1016/j.canlet.2021.01.014
|
| [40] |
L. G. de Pillis, A. Eladdadi, A. E. Radunskaya, Modeling cancer-immune responses to therapy, J. Pharmacokinet. Pharmacodyn., 41 (2014), 461–478. https://doi.org/10.1007/s10928-014-9386-9 doi: 10.1007/s10928-014-9386-9
|
| [41] |
R. Walker, H. Enderling, From concept to clinic: mathematically informed immunotherapy, Curr. Probl. Cancer, 40 (2016), 68–83. https://doi.org/10.1016/j.currproblcancer.2015.10.004 doi: 10.1016/j.currproblcancer.2015.10.004
|
| [42] |
A. Konstorum, A. T. Vella, A. J. Adler, R. C. Laubenbacher, Addressing current challenges in cancer immunotherapy with mathematical and computational modelling, J. R. Soc. Interface, 14 (2017), 20170150. https://doi.org/10.1098/rsif.2017.0150 doi: 10.1098/rsif.2017.0150
|
| [43] | U. Nukala, M. R. Messan, O. N. Yogurtcu, X. Wang, H. Yang, A systematic review of the efforts and hindrances of modeling and simulation of CAR T-cell therapy, AAPS J., 23 (2021), 1–20. |
| [44] |
L. R. C. Barros, E. A. Paixão, A. M. P. Valli, G. T. Naozuka, A. C. Fassoni, R. C. Almeida, CARTmath-a mathematical model of CAR-T tmmunotherapy in preclinical studies of hematological cancers, Cancers, 13 (2021), 2941. https://doi.org/10.3390/cancers13122941 doi: 10.3390/cancers13122941
|
| [45] |
O. León-Triana, A. Pérez-Martínez, M. Ramírez-Orellana, V. P. érez-García, Dual-target CAR-Ts with on-and off-tumour activity may override immune suppression in solid cancers: a mathematical proof of concept, Cancers, 13 (2021), 703. https://doi.org/10.3390/cancers13040703 doi: 10.3390/cancers13040703
|
| [46] | A. Chaudhury, X. Zhu, L. Chu, A. Goliaei, C. H. June, J. D. Kearns, et al., Chimeric antigen receptor T cell therapies: a review of cellular kinetic-pharmacodynamic modeling approaches, J. Clin. Pharmacol., 60 (2020), S147–S159. |
| [47] |
P. Sahoo, X. Yang, D. Abler, D. Maestrini, V. Adhikarla, D. Frankhouser, et al., Mathematical deconvolution of CAR T-cell proliferation and exhaustion from real-time killing assay data, J. R. Soc. Interface, 17 (2020), 20190734. https://doi.org/10.1098/rsif.2019.0734 doi: 10.1098/rsif.2019.0734
|
| [48] | K. Owens, I. Bozic, Modeling CAR T-cell therapy with patient preconditioning, Bull. Math. Biol., 83 (2021), 1–36. |
| [49] | Á. Martínez-Rubio, S. Chulián, C. B. Goñi, M. R. Orellana, A. P. Martínez, A. Navarro-Zapata, et al., A mathematical description of the bone marrow dynamics during CAR T-cell therapy in B-cell childhood acute lymphoblastic leukemia, Int. J. Mol. Sci., 22 (2021), 6371. |
| [50] |
K. J. Mahasa, A. Eladdadi, L. de Pillis, R. Ouifki, Oncolytic potency and reduced virus tumor-specificity in oncolytic virotherapy. A mathematical modelling approach, PLoS One, 12 (2017), e0184347. https://doi.org/10.1371/journal.pone.0184347 doi: 10.1371/journal.pone.0184347
|
| [51] |
K. M. Storey, S. E. Lawler, T. L. Jackson, Modeling oncolytic viral therapy, immune checkpoint inhibition, and the complex dynamics of innate and adaptive immunity in glioblastoma treatment, Front. Phys., 11 (2020), 151. https://doi.org/10.3389/fphys.2020.00151 doi: 10.3389/fphys.2020.00151
|
| [52] |
G. V. R. K. Vithanage, H. C. Wei, S. R. J. Jang, Bistability in a model of tumor-immune system interactions with an oncolytic viral therapy, Math. Biosci. Eng., 19 (2021), 1559–1587. https://doi.org/10.3934/mbe.2022072 doi: 10.3934/mbe.2022072
|
| [53] |
J. Malinzi, R. Ouifki, A. Eladdadi, D. F. M. Torres, K. A. J. White, Enhancement of chemotherapy using oncolytic virotherapy: Mathematical and optimal control analysis, Math. Biosci. Eng., 15 (2018), 1435–1463. https://doi.org/10.3934/mbe.2018066 doi: 10.3934/mbe.2018066
|
| [54] |
A. Diouf, H. Mokrani, E. Afenya, B. I. Camara, Computation of the conditions for anti-angiogenesis and gene therapy synergistic effects: Sensitivity analysis and robustness of target solutions, J. Theor. Biol., 528 (2021), 110850, https://doi.org/10.1016/j.jtbi.2021.110850 doi: 10.1016/j.jtbi.2021.110850
|
| [55] | S. R. J. Jang, H. C. Wei, On a mathematical model of tumor-immune system interactions with an oncolytic virus therapy, Discrete Contin. Dyn. Syst. B, 2021. |
| [56] |
R. Walker, P. E. Navas, S. H. Friedman, S. Galliani, A. Karolak, F. Macfarlane, et al., Enhancing synergy of CAR T cell therapy and oncolytic virus therapy for pancreatic cancer, bioRxiv, (2016), 055988. https://doi.org/10.1101/055988 doi: 10.1101/055988
|
| [57] | D. Wodarz, Viruses as antitumor weapons: defining conditions for tumor remission, Cancer Res., 61 (2001), 3501–3507. |
| [58] | A. L. Jenner, C. O. Yun, A. Yoon, A. C. F. Coster, P. S. Kim, Modelling combined virotherapy and immunotherapy: strengthening the antitumour immune response mediated by IL-$12$ and GM-CSF expression, Lett. Biomath., 5 (2018), S99–S116. |
| [59] |
N. Almuallem, D. Trucu, R. Eftimie, Oncolytic viral therapies and the delicate balance between virus-macrophage-tumour interactions: A mathematical approach, Math. Biosci. Eng., 18 (2021), 764–799. https://doi.org/10.3934/mbe.2021041 doi: 10.3934/mbe.2021041
|
| [60] |
A. Friedman, X. Lai, Combination therapy for cancer with oncolytic virus and checkpoint inhibitor: A mathematical model, PLoS One, 13 (2018), e0192449. https://doi.org/10.1371/journal.pone.0192449 doi: 10.1371/journal.pone.0192449
|
| [61] |
I. A. Rodriguez-Brenes, A. Hofacre, H. Fan, D. Wodarz, Complex dynamics of virus spread from low infection multiplicities: Implications for the spread of oncolytic viruses, PLoS Comput. Biol., 13 (2017), e1005241. https://doi.org/10.1371/journal.pcbi.1005241 doi: 10.1371/journal.pcbi.1005241
|
| [62] | L. R. Paiva, C. Binny, S. C. Ferreira, M. L. Martins, A multiscale mathematical model for oncolytic virotherapy, Cancer Res., 39 (2009), 1205–1211. |
| [63] |
D. Wodarz, A. Hofacre, J. W. Lau, Z. Sun, H. Fan, N. L. Komarova, Complex spatial dynamics of oncolytic viruses in vitro: mathematical and experimental approaches, PLoS Comput. Biol., 8 (2012), e1002547. https://doi.org/10.1371/journal.pcbi.1002547 doi: 10.1371/journal.pcbi.1002547
|
| [64] |
A. L. Jenner, A. C. F. Coster, P. S. Kim, F. Frascoli, Treating cancerous cells with viruses: insights from a minimal model for oncolytic virotherapy, Lett. Biomath., 5 (2018), S117–S136. https://doi.org/10.30707/LiB5.2Jenner doi: 10.30707/LiB5.2Jenner
|
| [65] | J. P. W. Heidbuechel, D. Abate-Daga, C. E. Engeland, H. Enderling, Mathematical modeling of oncolytic virotherapy, in Oncolytic viruses, methods in molecular biology, Springer, Humana, New York, (2020), 301–320. |
| [66] |
N. S. Senekal, K. J. Mahasa, A. Eladdadi, L. de Pillis, R. Ouifki, Natural killer cells recruitment in oncolytic virotherapy: a mathematical model, Bull. Math. Biol., 83 (2021), 1–51. https://doi.org/10.1007/s11538-021-00903-6 doi: 10.1007/s11538-021-00903-6
|
| [67] | J. King, K. S. Eroumé, R. Truckenmüller, S. Giselbrecht, A. E. Cowan, L. Loew, Ten steps to investigate a cellular system with mathematical modeling, PLoS Comput. Biol., 17 (2021), e1008921. |
| [68] |
L. G. de Pillis, A. E. Radunskaya, C. L. Wiseman, A validated mathematical model of cell-mediated immune response to tumour growth, Cancer Res., 65 (2005), 7950–7958. https://doi.org/10.1158/0008-5472.CAN-05-0564 doi: 10.1158/0008-5472.CAN-05-0564
|
| [69] |
D. Phan, D. Wodarz, Modeling multiple infection of cells by viruses: challenges and insights, Math. Biosci., 264 (2015), 21–28. https://doi.org/10.1016/j.mbs.2015.03.001 doi: 10.1016/j.mbs.2015.03.001
|
| [70] |
C. Grassberger, D. McClatchy, C. Geng, S. C. Kamran, F. Fintelmann, Y. E. Maruvka, et al., Patient-specific tumor growth trajectories determine persistent and resistant cancer cell populations during treatment with targeted therapies, Cancer Res., 79 (2019), 3776–3788. https://doi.org/10.1158/0008-5472.CAN-18-3652 doi: 10.1158/0008-5472.CAN-18-3652
|
| [71] |
A. L. Jenner, T. Cassidy, K. Belaid, M. C. Bourgeois-Daigneault, M. Craig, In silico trials predict that combination strategies for enhancing vesicular stomatitis oncolytic virus are determined by tumor aggressivity, J. Immunother. Cancer, 9 (2021), e001387. https://doi.org/10.1136/jitc-2020-001387 doi: 10.1136/jitc-2020-001387
|
| [72] | H. Enderling, O. Wolkenhauer, Are all models wrong? Comput. Syst., 1 (2020), e1008. https: //doi.org/10.1002/cso2.1008 |
| [73] |
P. S. Kim, J. J. Crivelli, I. K. Choi, C. O. Yun, J. R. Wares, Quantitative impact of immunomodulation versus oncolysis with cytokine-expressing virus therapeutics, Math. Biosci. Eng., 12 (2015), 841–858. https://doi.org/10.1002/cbdv.201400024 doi: 10.1002/cbdv.201400024
|
| [74] |
J. J. Crivelli, J. Földes, P. S. Kim, J. R. Wares, A mathematical model for cell cycle-specific cancer virotherapy, J. Biol. Dyn., 6 (2012), 104–120. https://doi.org/10.1080/17513758.2011.613486 doi: 10.1080/17513758.2011.613486
|
| [75] |
A. L. Jenner, P. S. Kim, F. Frascoli, Oncolytic virotherapy for tumours following a Gompertz growth law, J. Theor. Biol., 480 (2019), 129–140. https://doi.org/10.1016/j.jtbi.2019.08.002 doi: 10.1016/j.jtbi.2019.08.002
|
| [76] |
H. Huang, Y. Liu, W. Liao, Y. Cao, Q. Liu, Y. Guo, et al., Oncolytic adenovirus programmed by synthetic gene circuit for cancer immunotherapy, Nat. Commun., 10 (2019), 1–15. https://doi.org/10.1038/s41467-019-12794-2 doi: 10.1038/s41467-019-12794-2
|
| [77] |
M. Ruella, M. Klichinsky, S. S. Kenderian, O. Shestova, A. Ziober, D. O. Kraft, et al., Overcoming the immunosuppressive tumor microenvironment of Hodgkin lymphoma using chimeric antigen receptor T cells, Cancer Discovery, 7 (2017), 1154–1167. https://doi.org/10.1158/2159-8290.CD-16-0850 doi: 10.1158/2159-8290.CD-16-0850
|
| [78] |
A. Friedman, J. P. Tian, G. Fulci, E. A. Chiocca, J. Wang, Glioma virotherapy: effects of innate immune suppression and increased viral replication capacity, Cancer Res., 66 (2006), 2314–2319. https://doi.org/10.1158/0008-5472.CAN-05-2661 doi: 10.1158/0008-5472.CAN-05-2661
|
| [79] |
R. Eftimie, C. K. Macnamara, J. Dushoff, J. L. Bramson, D. J. Earn, Bifurcations and chaotic dynamics in a tumour-immune-virus system, Math. Modell. Nat. Phenom., 11 (2016), 65–85. https://doi.org/10.1051/mmnp/201611505 doi: 10.1051/mmnp/201611505
|
| [80] |
R. Eftimie, G. Eftimie, Investigating macrophages plasticity following tumour–immune interactions during oncolytic therapies, Acta Biotheor., 67 (2019), 321–359. https://doi.org/10.1007/s10441-019-09357-9 doi: 10.1007/s10441-019-09357-9
|
| [81] |
Ž Bajzer, T. Carr, K. Josić, S. J. Russell, D. Dingli, Modeling of cancer virotherapy with recombinant measles viruses, J. Theor. Biol., 252 (2008), 109–122. https://doi.org/10.1016/j.jtbi.2008.01.016 doi: 10.1016/j.jtbi.2008.01.016
|
| [82] |
K. Jacobsen, L. Russell, B. Kaur, A. Friedman, Effects of CCN1 and macrophage content on glioma virotherapy: a mathematical model, Bull. Math. Biol., 77 (2015), 1–29. https://doi.org/10.1007/s11538-014-0046-4 doi: 10.1007/s11538-014-0046-4
|
| [83] |
M. Aghi, R. L. Martuza, Oncolytic viral therapies–the clinical experience, Oncogene, 24 (2005), 7802–7816. https://doi.org/10.1038/sj.onc.1209037 doi: 10.1038/sj.onc.1209037
|
| [84] | A. Saltelli, M. Ratto, T. Andres, F. Campolongo, J. Cariboni, D. Gatelli, et al., Global Sensitivity Analysis: the Primer, Wiley, New York, 2008. |
| [85] |
B. Niu, X. Zeng, T. A. Phan, F. Szulzewsky, S. Holte, E. C. Holland, et al., Mathematical modeling of PDGF-driven glioma reveals the dynamics of immune cells infiltrating into tumors, Neoplasia, 22 (2020), 323–332. https://doi.org/10.1016/j.neo.2020.05.005 doi: 10.1016/j.neo.2020.05.005
|
| [86] |
A. L. Jenner, C. O. Yun, P. S. Kim, A. C. F. Coster, Mathematical modelling of the interaction between cancer cells and an oncolytic virus: insights into the effects of treatment protocols, Bull. Math. Biol., 80 (2018), 1615–1629. https://doi.org/10.1007/s11538-018-0424-4 doi: 10.1007/s11538-018-0424-4
|
| [87] |
T. Krabbe, J. Marek, T. Groll, K. Steiger, R. M. Schmid, A. M. Krackhardt, et al., Adoptive T cell therapy is complemented by oncolytic virotherapy with fusogenic VSV-NDV in combination treatment of murine melanoma, Cancers, 13 (2021), 1044. https://doi.org/10.3390/cancers13051044 doi: 10.3390/cancers13051044
|
| [88] |
Z. P. Parra-Guillen, T. Freshwater, Y. Cao, K. Mayawala, S. Zalba, M. J. Garrido, et al., Mechanistic modeling of a novel oncolytic virus, V937, to describe viral kinetic and dynamic processes following intratumoral and intravenous administration, Front. Pharmacol., 12 (2021), 705443. https://doi.org/10.3389/fphar.2021.705443 doi: 10.3389/fphar.2021.705443
|
| [89] |
A. Mueller-Schoell, N. Puebla-Osorio, R. Michelet, M. R. Green, A. Künkele, W. Huisinga, et al., Early survival prediction framework in CD19-specific CAR-T cell immunotherapy using a quantitative systems pharmacology model, Cancers, 13 (2021), 2782. https://doi.org/10.3390/cancers13112782 doi: 10.3390/cancers13112782
|
| [90] |
G. V. R. K. Vithanage, H. C. Wei, S. R. J. Jang, Bistability in a model of tumor-immune system interactions with an oncolytic viral therapy, Math. Biosci. Eng., 19 (2021), 1559–1587. https://doi.org/10.3934/mbe.2022072 doi: 10.3934/mbe.2022072
|
| [91] |
A. Takasu, A. Masui, M. Hamada, T. Imai, S. Iwai, Y. Yura, Immunogenic cell death by oncolytic herpes simplex virus type 1 in squamous cell carcinoma cells, Cancer Gene Ther., 23 (2016), 107–113. https://doi.org/10.5194/npg-23-107-2016 doi: 10.5194/npg-23-107-2016
|
| [92] |
P. K. Bommareddy, A. Zloza, S. D. Rabkin, H. L. Kaufman, Oncolytic virus immunotherapy induces immunogenic cell death and overcomes STING deficiency in melanoma, OncoImmunology, 8 (2019), e1591875. https://doi.org/10.1080/2162402X.2019.1591875 doi: 10.1080/2162402X.2019.1591875
|
| [93] |
J. P. van Vloten, S. T. Workenhe, S. K. Wootton, K. L. Mossman, B. W. Bridle, Critical interactions between immunogenic cancer cell death, oncolytic viruses, and the immune system define the rational design of combination immunotherapies, J. Immunol., 200 (2018), 450–458. https://doi.org/10.4049/jimmunol.1701021 doi: 10.4049/jimmunol.1701021
|
| [94] |
D. Wodarz, N. Komarova, Towards predictive computational models of oncolytic virus therapy: basis for experimental validation and model selection, PLoS One, 4 (2009), e4271. https://doi.org/10.1371/journal.pone.0004271 doi: 10.1371/journal.pone.0004271
|
| [95] |
T. J. Paul, The replicability of oncolytic virus: defining conditions in tumor virotherapy, Math. Biosci. Eng., 8 (2011), 841–860. https://doi.org/10.3934/mbe.2011.8.841 doi: 10.3934/mbe.2011.8.841
|
| [96] |
P. Pooladvand, C. O. Yun, A. R. Yoon, P. S. Kim, F. Frascoli, The role of viral infectivity in oncolytic virotherapy outcomes: A mathematical study, Math. Biosci., 334 (2021), 108520. https://doi.org/10.1016/j.mbs.2020.108520 doi: 10.1016/j.mbs.2020.108520
|
| [97] |
S. T. Workenhe, G. Simmons, J. G. Pol, B. D. Lichty, W. P. Halford, K. L. Mossman, Immunogenic HSV-mediated oncolysis shapes the antitumor immune response and contributes to therapeutic efficacy, Mol. Ther., 22 (2014), 123–131. https://doi.org/10.1038/mt.2013.238 doi: 10.1038/mt.2013.238
|
| [98] |
O. J. Kwon, E. Kang, S. Kim, C. O. Yun, Viral genome DNA/lipoplexes elicit in situ oncolytic viral replication and potent antitumor efficacy via systemic delivery, J. Controlled Release, 155 (2011), 317–325. https://doi.org/10.1016/j.jconrel.2011.06.014 doi: 10.1016/j.jconrel.2011.06.014
|
| [99] |
A. Mohamed, R. N. Johnston, M. Shmulevitz, Potential for improving potency and specificity of reovirus oncolysis with next-generation reovirus variants, Viruses, 7 (2015), 6251–6278. https://doi.org/10.3390/v7122936 doi: 10.3390/v7122936
|
| [100] |
J. Altomonte, L. Wu, M. Meseck, L. Chen, O. Ebert, A. Garcia-Sastre, et al., Enhanced oncolytic potency of vesicular stomatitis virus through vector-mediated inhibition of NK and NKT cells, Cancer Gene Ther., 16 (2009), 266–278. https://doi.org/10.1038/cgt.2008.74 doi: 10.1038/cgt.2008.74
|
| [101] |
C. A. Alvarez-Breckenridge, J. Yu, R. Price, J. Wojton, J. Pradarelli, H. Mao, et al., NK cells impede glioblastoma virotherapy through NKp30 and NKp46 natural cytotoxicity receptors, Nat. Med., 18 (2012), 1827–1834. https://doi.org/10.1038/nm.3013 doi: 10.1038/nm.3013
|
| [102] |
A. L. de Matos, L. S. Franco, G. McFadden, Oncolytic viruses and the immune system: the dynamic duo, Mol. Ther. Methods Clin. Dev., 17 (2020), 349–358. https://doi.org/10.1016/j.omtm.2020.01.001 doi: 10.1016/j.omtm.2020.01.001
|
| [103] |
F. Marofi, R. Motavalli, V. A. Safonov, L. Thangavelu, A. V. Yumashev, M. Alexander, et al., CAR T cells in solid tumors: challenges and opportunities, Stem Cell Res. Ther., 12 (2021), 1–16. https://doi.org/10.1186/s13287-020-02006-w doi: 10.1186/s13287-020-02006-w
|
| [104] |
S. Kailayangiri, B. Altvater, M. Wiebel, S. Jamitzky, Overcoming heterogeneity of antigen expression for effective CAR T cell targeting of cancers, Cancers, 12 (2020), 1075. https://doi.org/10.3390/cancers12051075 doi: 10.3390/cancers12051075
|
| [105] | N. V. Frey, P. A. Shaw, E. O. Hexner, S. Gill, K. Marcucci, S. M. Luger, et al., Optimizing chimeric antigen receptor (CAR) T cell therapy for adult patients with relapsed or refractory (r/r) acute lymphoblastic leukemia (ALL), J. Clin. Oncol., 34 (2016), 7002. |
| [106] |
J. P. W. Heidbuechel, C. E. Engeland, Oncolytic viruses encoding bispecific T cell engagers: a blueprint for emerging immunovirotherapies, J. Hematol. Oncol., 14 (2021), 1–24. https://doi.org/10.1186/s13045-021-01075-5 doi: 10.1186/s13045-021-01075-5
|
| [107] |
A. Wing, C. A. Fajardo, A. D. Posey, C. Shaw, T. Da, R. M. Young, et al., Improving CART-cell therapy of solid tumors with oncolytic virus–driven production of a bispecific T-cell engager, Cancer Immunol. Res., 6 (2018), 605–616. https://doi.org/10.1158/2326-6066.CIR-17-0314 doi: 10.1158/2326-6066.CIR-17-0314
|
| [108] |
O. P. Nave, M. Elbaz, S. Bunimovich-Mendrazitsky, Analysis of a breast cancer mathematical model by a new method to find an optimal protocol for HER2-positive cancer, Biosystems, 197 (2020), 104191. https://doi.org/10.1016/j.biosystems.2020.104191 doi: 10.1016/j.biosystems.2020.104191
|
| [109] |
O. P. Nave, S. Hareli, M. Elbaz, I. H. Iluz, S. Bunimovich-Mendrazitsky, BCG and IL - 2 model for bladder cancer treatment with fast and slow dynamics based on SPVF method–stability analysis, Math. Biosci. Eng., 16 (2019), 5346–5379. https://doi.org/10.3934/mbe.2019267 doi: 10.3934/mbe.2019267
|
| [110] |
S. Marino, I. B. Hogue, C. J. Ray, D. E. Kirschner, A methodology for performing global uncertainty and sensitivity analysis in systems biology, J. Theor. Biol., 254 (2008), 178–196. https://doi.org/10.1016/j.jtbi.2008.04.011 doi: 10.1016/j.jtbi.2008.04.011
|













Figures(14) / Tables(1)

Khaphetsi Joseph Mahasa, Rachid Ouifki, Amina Eladdadi, Lisette de Pillis. A combination therapy of oncolytic viruses and chimeric antigen receptor T cells: a mathematical model proof-of-concept[J]. Mathematical Biosciences and Engineering, 2022, 19(5): 4429-4457. doi: 10.3934/mbe.2022205







 DownLoad:
DownLoad: