We aimed to construct a novel prognostic model based on N6-methyladenosine (m6A)-related autophagy genes for predicting the prognosis of lung squamous cell carcinoma (LUSC).
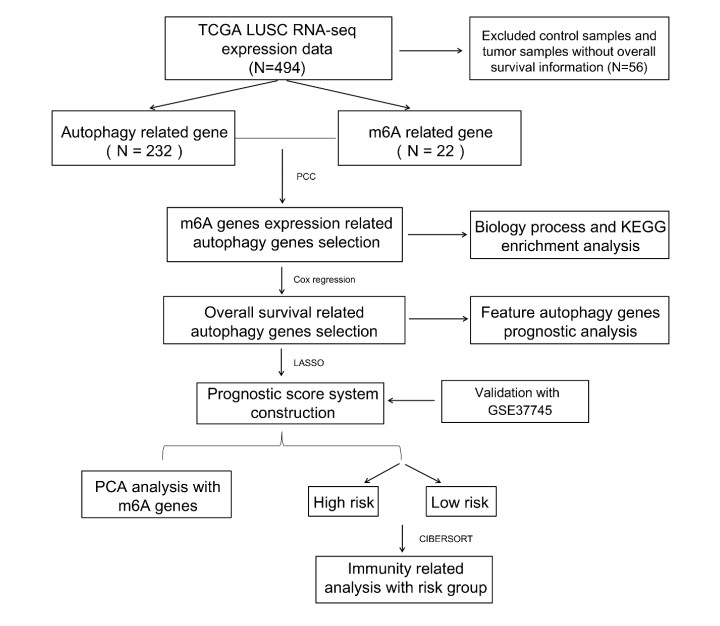
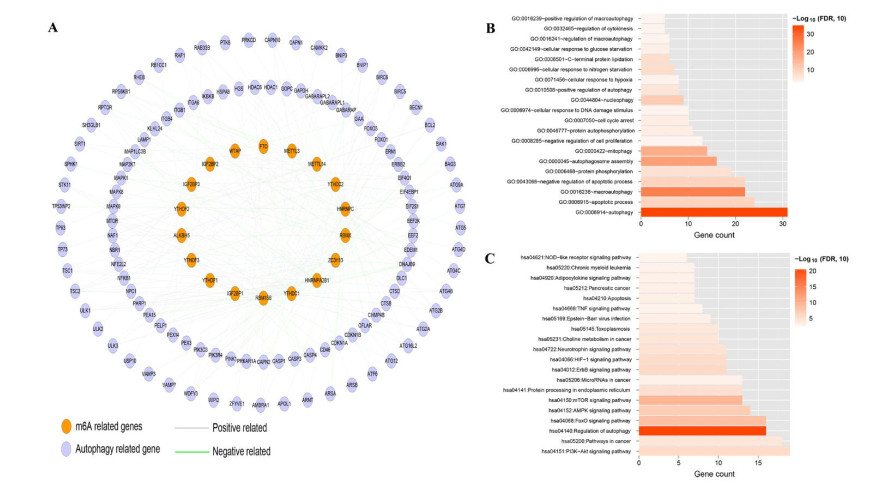
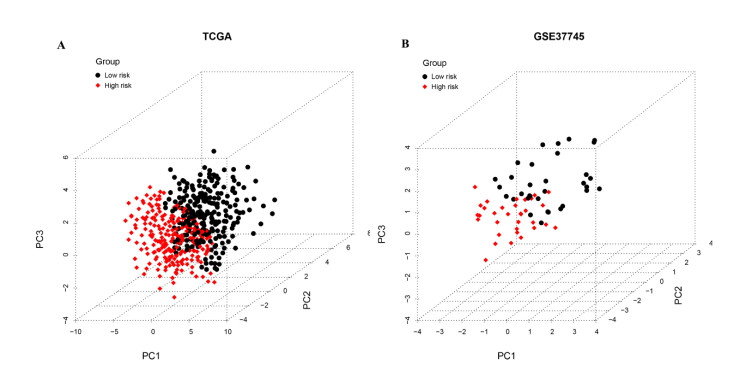
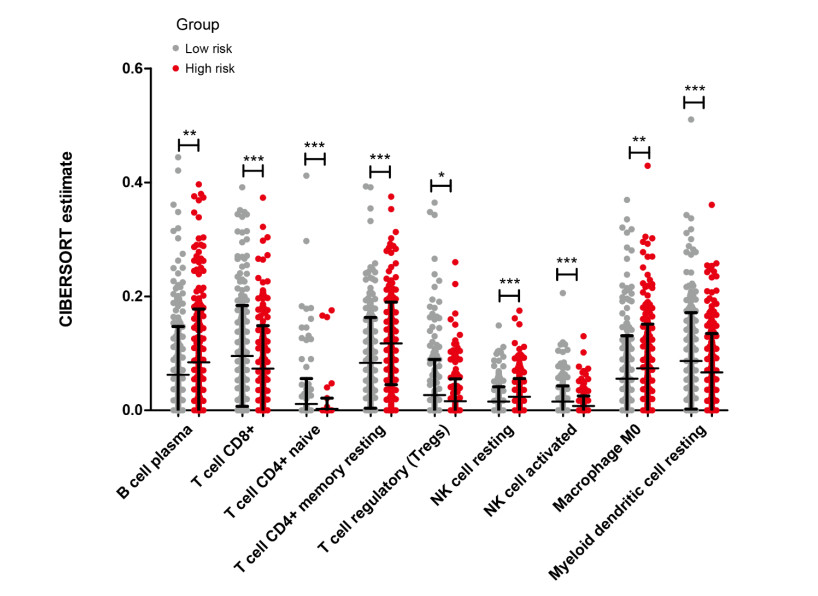
Gene expression profiles and clinical information of Patients with LUSC were downloaded from The Cancer Genome Atlas (TCGA) database. In addition, m6A- and autophagy-related gene profiles were obtained from TCGA and Human Autophagy Database, respectively. Pearson correlation analysis was performed to identify the m6A-related autophagy genes, and univariate Cox regression analysis was conducted to screen for genes associated with prognosis. Based on these genes, LASSO Cox regression analysis was used to construct a prognostic model. The corresponding prognostic score (PS) was calculated, and patients with LUSC were assigned to low- and high-risk groups according to the median PS value. An independent dataset (GSE37745) was used to validate the prognostic ability of the model. CIBERSORT was used to calculate the differences in immune cell infiltration between the high- and low-risk groups.
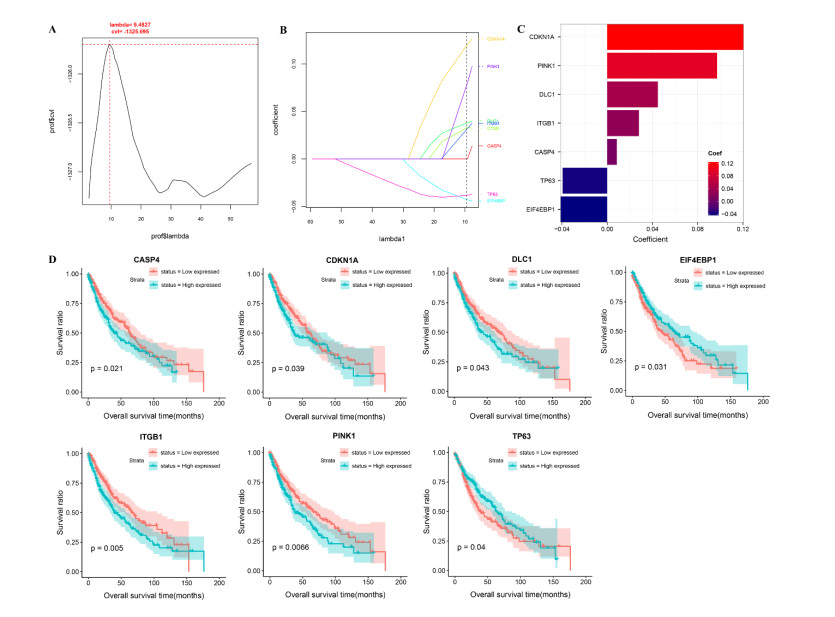
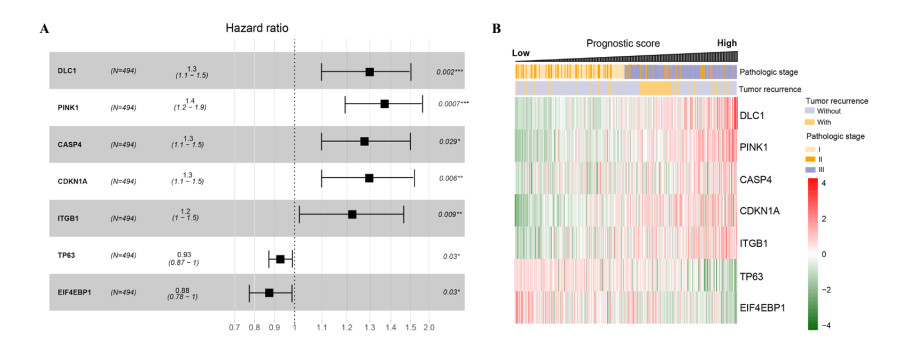
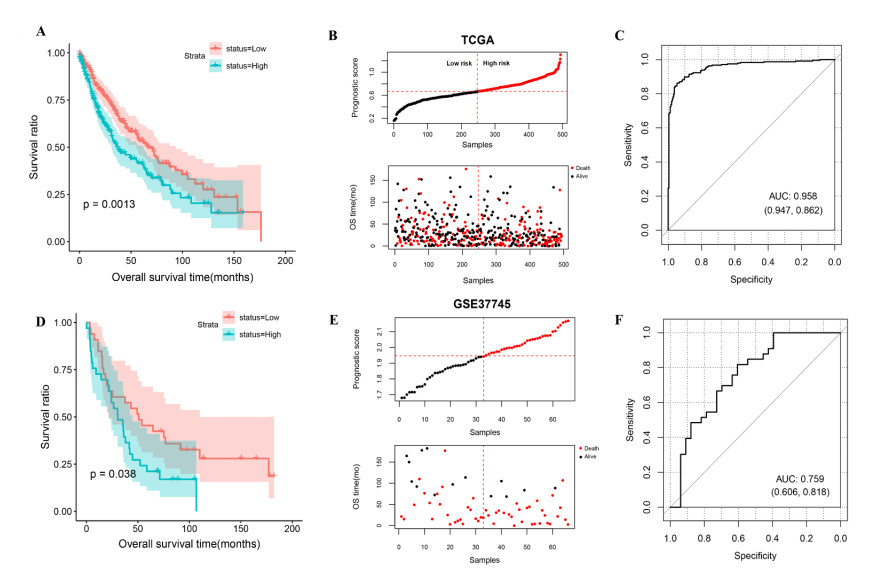
Seven m6A-related autophagy genes were screened to construct a prognostic model: CASP4, CDKN1A, DLC1, ITGB1, PINK1, TP63, and EIF4EBP1. In the training and validation sets, patients in the high-risk group had worse survival times than those in the low-risk group; the areas under the receiver operating characteristic curves were 0.958 and 0.759, respectively. There were differences in m6A levels and immune cell infiltration between the high- and low-risk groups.
Our prognostic model of the seven m6A-related autophagy genes had significant predictive value for LUSC; thus, these genes may serve as autophagy-related therapeutic targets in clinical practice.
Citation: Xin Yu, Jun Liu, Ruiwen Xie, Mengling Chang, Bichun Xu, Yangqing Zhu, Yuancai Xie, Shengli Yang. Construction of a prognostic model for lung squamous cell carcinoma based on seven N6-methylandenosine-related autophagy genes[J]. Mathematical Biosciences and Engineering, 2021, 18(5): 6709-6723. doi: 10.3934/mbe.2021333
We aimed to construct a novel prognostic model based on N6-methyladenosine (m6A)-related autophagy genes for predicting the prognosis of lung squamous cell carcinoma (LUSC).
Gene expression profiles and clinical information of Patients with LUSC were downloaded from The Cancer Genome Atlas (TCGA) database. In addition, m6A- and autophagy-related gene profiles were obtained from TCGA and Human Autophagy Database, respectively. Pearson correlation analysis was performed to identify the m6A-related autophagy genes, and univariate Cox regression analysis was conducted to screen for genes associated with prognosis. Based on these genes, LASSO Cox regression analysis was used to construct a prognostic model. The corresponding prognostic score (PS) was calculated, and patients with LUSC were assigned to low- and high-risk groups according to the median PS value. An independent dataset (GSE37745) was used to validate the prognostic ability of the model. CIBERSORT was used to calculate the differences in immune cell infiltration between the high- and low-risk groups.
Seven m6A-related autophagy genes were screened to construct a prognostic model: CASP4, CDKN1A, DLC1, ITGB1, PINK1, TP63, and EIF4EBP1. In the training and validation sets, patients in the high-risk group had worse survival times than those in the low-risk group; the areas under the receiver operating characteristic curves were 0.958 and 0.759, respectively. There were differences in m6A levels and immune cell infiltration between the high- and low-risk groups.
Our prognostic model of the seven m6A-related autophagy genes had significant predictive value for LUSC; thus, these genes may serve as autophagy-related therapeutic targets in clinical practice.

| [1] |
A. Friedlaender, A. Addeo, A. Russo, V. Gregorc, D. Cortinovis, C. D. Rolfo, Targeted therapies in early stage NSCLC: Hype or hope?, Int. J. Mol. Sci., 21 (2020), 6329. doi: 10.3390/ijms21176329

|
| [2] |
M. Gao, W. Kong, Z. Huang, Z. Xie, Identification of key genes related to lung squamous cell carcinoma using bioinformatics analysis, Int. J. Mol. Sci., 21 (2020), 2994. doi: 10.3390/ijms21082994
|
| [3] |
N. Mizushima, B. Levine, A. M. Cuervo, D. J. Klionsky, Autophagy fights disease through cellular self-digestion, Nature, 451 (2008), 1069–1075. doi: 10.1038/nature06639
|
| [4] |
R. F. Zaarour, B. Azakir, E. Y. Hajam, H. Nawafleh, N. A. Zeinelabdin, A. S. T. Engelsen, et al., Role of hypoxia-mediated autophagy in tumor cell death and survival, Cancers (Basel), 13 (2021), 533. doi: 10.3390/cancers13030533
|
| [5] |
Z. Yang, D. J. Klionsky, Mammalian autophagy: core molecular machinery and signaling regulation, Curr. Opin.Cell Biol., 22 (2010), 124–131. doi: 10.1016/j.ceb.2009.11.014
|
| [6] | T. Losmanová, F. A. Janser, M. Humbert, I. Tokarchuk, A. M. Schläfli, C. Neppl, et al., Chaperone-mediated autophagy markers LAMP2A and HSC70 are independent adverse prognostic markers in primary resected squamous cell carcinomas of the lung, Oxidat. Med. Cellul. Longev., 2020 (2020), 8506572. |
| [7] | W. Xu, B. Chen, D. Ke, X. Chen, TRIM29 mediates lung squamous cell carcinoma cell metastasis by regulating autophagic degradation of E-cadherin, Aging (Albany NY), 12 (2020), 13488–13501. |
| [8] |
L. He, H. Li, A. Wu, Y. Peng, G. Shu, G. Yin, Functions of N6-methyladenosine and its role in cancer, Mol. Cancer, 18 (2019), 176. doi: 10.1186/s12943-019-1109-9
|
| [9] |
J. Yang, J. Chen, X. Fei, X. Wang, K. Wang, N6-methyladenine RNA modification and cancer, Oncol. Lett., 20 (2020), 1504–1512. doi: 10.3892/ol.2020.11739
|
| [10] |
Y. Liu, X. Guo, M. Zhao, H. Ao, X. Leng, M. Liu, et al., Contributions and prognostic values of m(6) A RNA methylation regulators in non-small-cell lung cancer, J. Cell Physiol., 235 (2020), 6043–6057. doi: 10.1002/jcp.29531
|
| [11] |
S. Sun, Q. Han, M. Liang, Q. Zhang, J. Zhang, J. Cao, Downregulation of m(6) A reader YTHDC2 promotes tumor progression and predicts poor prognosis in non-small cell lung cancer, Thorac. Cancer, 11 (2020), 3269–3279. doi: 10.1111/1759-7714.13667
|
| [12] |
J. Botling, K. Edlund, M. Lohr, B. Hellwig, L. Holmberg, M. Lambe, et al., Biomarker discovery in non-small cell lung cancer: integrating gene expression profiling, meta-analysis, and tissue microarray validation, Clin. Cancer Res., 19 (2013), 194–204. doi: 10.1158/1078-0432.CCR-12-1139
|
| [13] |
V. Jabs, K. Edlund, H. Konig, M. Grinberg, K. Madjar, J. Rahnenfuhrer, et al., Integrative analysis of genome-wide gene copy number changes and gene expression in non-small cell lung cancer, PLoS One, 12 (2017), e0187246. doi: 10.1371/journal.pone.0187246
|
| [14] |
M. Lohr, B. Hellwig, K. Edlund, J. S. Mattsson, J. Botling, M. Schmidt, et al., Identification of sample annotation errors in gene expression datasets, Arch. Toxicol., 89 (2015), 2265–2272. doi: 10.1007/s00204-015-1632-4
|
| [15] |
P. Shannon, A. Markiel, O. Ozier, N. S. Baliga, J. T. Wang, D. Ramage, et al., Cytoscape: a software environment for integrated models of biomolecular interaction networks, Genome Res., 13 (2003), 2498–2504. doi: 10.1101/gr.1239303
|
| [16] |
W. Huang da, B. T. Sherman, R. A. Lempicki, Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources, Nat. Protoc., 4 (2009), 44–57. doi: 10.1038/nprot.2008.211
|
| [17] |
W. Huang da, B. T. Sherman, R. A. Lempicki, Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists, Nucleic Acids Res., 37 (2009), 1–13. doi: 10.1093/nar/gkn923
|
| [18] |
P. Wang, Y. Wang, B. Hang, X. Zou, J. H. Mao, A novel gene expression-based prognostic scoring system to predict survival in gastric cancer, Oncotarget, 7 (2016), 55343–55351. doi: 10.18632/oncotarget.10533
|
| [19] |
R. Tibshirani, The lasso method for variable selection in the Cox model, Stat. Med., 16 (1997), 385–395. doi: 10.1002/(SICI)1097-0258(19970228)16:4<385::AID-SIM380>3.0.CO;2-3
|
| [20] | J. J. Goeman, L1 penalized estimation in the Cox proportional hazards model, Biom. J., 52 (2010), 70–84. |
| [21] |
E. Alizadeh, S. M. Lyons, J. M. Castle, A. Prasad, Measuring systematic changes in invasive cancer cell shape using Zernike moments, Integr. Biol. (Camb), 8 (2016), 1183–1193. doi: 10.1039/C6IB00100A
|
| [22] |
U. Sinha, H. Kangarloo, Principal component analysis for content-based image retrieval, Radiographics, 22 (2002), 1271–1289. doi: 10.1148/radiographics.22.5.g02se021271
|
| [23] |
B. C. Bade, C. S. D. Cruz, Lung cancer 2020: Epidemiology, etiology and prevention, Clin. Chest Med., 41 (2020), 1–24. doi: 10.1016/j.ccm.2019.10.001
|
| [24] | C. Gu, X. Shi, C. Dai, F. Shen, G. Rocco, J. Chen, et al., RNA m 6 A modification in cancers: Molecular mechanisms and potential clinical applications, Innovation, 1 (2020), 100066. |
| [25] |
E. White, J. M. Mehnert, C. S. Chan, Autophagy, metabolism and cancer, Clin. Cancer Res., 21 (2015), 5037–5046. doi: 10.1158/1078-0432.CCR-15-0490
|
| [26] |
X. Wang, R. Wu, Y. Liu, Y. Zhao, Z. Bi, Y. Yao, et al., m(6)A mRNA methylation controls autophagy and adipogenesis by targeting Atg5 and Atg7, Autophagy, 16 (2020), 1221–1235. doi: 10.1080/15548627.2019.1659617
|
| [27] |
J. T. Beck, A. Ismail, C. Tolomeo, Targeting the phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway: an emerging treatment strategy for squamous cell lung carcinoma, Cancer Treat. Rev., 40 (2014), 980–989. doi: 10.1016/j.ctrv.2014.06.006
|
| [28] |
Z. Sun, Z. Wang, X. Liu, D. Wang, New development of inhibitors targeting the PI3K/AKT/mTOR pathway in personalized treatment of non-small-cell lung cancer, Anticancer Drugs, 26 (2015), 1–14. doi: 10.1097/CAD.0000000000000172
|
| [29] |
N. Kayagaki, S. Warming, M. Lamkanfi, L. Vande Walle, S. Louie, J. Dong, et al., Non-canonical inflammasome activation targets caspase-11, Nature, 479 (2011), 117–121. doi: 10.1038/nature10558
|
| [30] |
M. Terlizzi, C. Colarusso, I. De Rosa, P. Somma, C. Curcio, R. P. Aquino, et al., Identification of a novel subpopulation of Caspase-4 positive non-small cell lung Cancer patients, J. Exp. Clin. Cancer Res. 39 (2020), 242. doi: 10.1186/s13046-020-01754-0
|
| [31] |
M. Zhao, C. Tong, Z. Hao, R. Zhao, L. Wang, MicroRNA-374b mediates the initiation of non-small cell lung cancer by regulating ITGB1 and p53 expressions, Thorac. Cancer, 11 (2020), 1670–1678. doi: 10.1111/1759-7714.13457
|
| [32] |
T. Fukazawa, M. Guo, N. Ishida, T. Yamatsuji, M. Takaoka, E. Yokota, et al., SOX2 suppresses CDKN1A to sustain growth of lung squamous cell carcinoma, Sci. Rep., 6 (2016), 20113. doi: 10.1038/srep20113
|
| [33] |
T. Y. Kim, S. Jackson, Y. Xiong, T. G. Whitsett, J. R. Lobello, G. J. Weiss, et al., CRL4A-FBXW5-mediated degradation of DLC1 Rho GTPase-activating protein tumor suppressor promotes non-small cell lung cancer cell growth, Proc. Nat. Acad. Sci. U. S. A., 110 (2013), 16868–16873. doi: 10.1073/pnas.1306358110
|
| [34] |
X. Lu, Q. X. Liu, J. Zhang, D. Zhou, G. X. Yang, M. Y. Li, et al., PINK1 overexpression promotes cell migration and proliferation via regulation of autophagy and predicts a poor prognosis in lung cancer cases, Cancer Manage. Res., 12 (2020), 7703–7714. doi: 10.2147/CMAR.S262466
|
| [35] |
B. Cao, P. Wang, L. Gu, J. Liu, Use of four genes in exosomes as biomarkers for the identification of lung adenocarcinoma and lung squamous cell carcinoma, Oncol. Lett., 21 (2021), 249. doi: 10.3892/ol.2021.12510
|
| [36] |
J. Zhu, M. Wang, D. Hu, Development of an autophagy-related gene prognostic signature in lung adenocarcinoma and lung squamous cell carcinoma, PeerJ, 8 (2020), e8288. doi: 10.7717/peerj.8288
|
| [37] |
X. Liu, S. Wu, Y. Yang, M. Zhao, G. Zhu, Z. Hou, The prognostic landscape of tumor-infiltrating immune cell and immunomodulators in lung cancer, Biomed. Pharmacother., 95 (2017), 55–61. doi: 10.1016/j.biopha.2017.08.003
|
| [38] |
Y. Zhu, X. Zhang, Investigating the significance of tumor-infiltrating immune cells for the prognosis of lung squamous cell carcinoma, PeerJ, 7 (2019), e7918. doi: 10.7717/peerj.7918
|
| [39] |
J. Li, H. Li, C. Zhang, C. Zhang, H. Wang, Integrative analysis of genomic alteration, immune cells infiltration and prognosis of lung squamous cell carcinoma (LUSC) to identify smoking-related biomarkers, Int. Immunopharmacol., 89 (2020), 107053. doi: 10.1016/j.intimp.2020.107053
|
| [40] |
Q. F. Yang, D. Wu, J. Wang, L. Ba, C. Tian, Y. T. Liu, et al., Development and validation of an individualized immune prognostic model in stage I-III lung squamous cell carcinoma, Sci. Rep., 11 (2021), 12727. doi: 10.1038/s41598-021-92115-0
|
| [41] |
A. B. Schulze, G. Evers, D. Görlich, M. Mohr, A. Marra, L. Hillejan, et al., Tumor infiltrating T cells influence prognosis in stage I-III non-small cell lung cancer, J. Thorac. Dis., 12 (2020), 1824–1842. doi: 10.21037/jtd-19-3414a
|
| [42] |
S. Jin, Y. Deng, J. W. Hao, Y. Li, B. Liu, Y. Yu, et al., NK cell phenotypic modulation in lung cancer environment, PLoS One, 9 (2014), e109976. doi: 10.1371/journal.pone.0086317
|
 mbe-18-05-333-Table S1-supplementary.xls mbe-18-05-333-Table S1-supplementary.xls |
 |
| mbe-18-05-333-Table S2-supplementary.xlsx |
|
| mbe-18-05-333-Table S3-supplementary.xlsx |
|
| mbe-18-05-333-Table S4-supplementary.xlsx |
|
Figures(7) / Tables(1)

Xin Yu, Jun Liu, Ruiwen Xie, Mengling Chang, Bichun Xu, Yangqing Zhu, Yuancai Xie, Shengli Yang. Construction of a prognostic model for lung squamous cell carcinoma based on seven N6-methylandenosine-related autophagy genes[J]. Mathematical Biosciences and Engineering, 2021, 18(5): 6709-6723. doi: 10.3934/mbe.2021333







 DownLoad:
DownLoad: