Citation: Mingzhan Huang, Shouzong Liu, Xinyu Song. Study of the sterile insect release technique for a two-sex mosquito population model[J]. Mathematical Biosciences and Engineering, 2021, 18(2): 1314-1339. doi: 10.3934/mbe.2021069

| [1] |
H. J. Barclay, The sterile insect release method on species with two-stage life cycles, Res. Popul. Ecol., 21 (1980), 165–180. doi: 10.1007/BF02513619

|
| [2] |
H. J. Barclay, M. Mackauer, The sterile insect release method for pest control: A density dependent model, Environ. Entomol., 9 (1980), 810–817. doi: 10.1093/ee/9.6.810
|
| [3] |
H. J. Barclay, Pest population stability under sterile releases, Res. Popul. Ecol., 24 (1982), 405–416. doi: 10.1007/BF02515585
|
| [4] |
H. J. Barclay, Modeling incomplete sterility in a sterile release program: Interactions with other factors, Popul. Ecol., 43 (2001), 197–206. doi: 10.1007/s10144-001-8183-7
|
| [5] | H. J. Barclay, Mathematical models for the use of sterile insects, in Sterile Insect Technique, Springer, Heidelberg, (2005), 147–174. |
| [6] |
L. Alphey, M. Benedict, R. Bellini, G. G. Clark, D. A. Dame, M. W. Service, et al., Sterile-insect methods for control of mosquito-borne diseases: an analysis, Vector-Borne Zoonotic Dis., 10 (2010), 295–311. doi: 10.1089/vbz.2009.0014
|
| [7] | W. Klassen, Area-wide integrated pest management and the sterile insect technique, in Sterile Insect Technique (eds. V. A. Dyck, J. Hendrichs and A. S. Robinson), Springer, The Netherlands, (2005), 39–68. |
| [8] |
M. Strugarek, H. Bossin, Y. Dumont, On the use of the sterile insect release technique to reduce or eliminate mosquito populations, Appl. Math. Model., 68 (2019), 443–470. doi: 10.1016/j.apm.2018.11.026
|
| [9] |
H. Laven, Eradication of culex pipiens fatigans through cytoplasmic incompatibility, Nature, 216 (1967), 383–384. doi: 10.1038/216383a0
|
| [10] |
X. Zheng, D. Zhang, Y. Li, C. Yang, Y. Wu, X. Liang, et al., Incompatible and sterile insect techniques combined eliminate mosquitoes, Nature, 572 (2019), 56–61. doi: 10.1038/s41586-019-1407-9
|
| [11] |
K. R. Fister, M. L. Mccarthy, S. F. Oppenheimer, C. Collins, Optimal control of insects through sterile insect release and habitat modification, Math. Biosci., 244 (2013), 201–212. doi: 10.1016/j.mbs.2013.05.008
|
| [12] |
S. M. White, P. Rohani, S. M. Sait, Modelling pulsed releases for sterile insect techniques: fitness costs of sterile and transgenic males and the effects on mosquito dynamics, J. Appl. Ecol., 47 (2010), 1329–1339. doi: 10.1111/j.1365-2664.2010.01880.x
|
| [13] | J. Li, Z. Yuan, Modeling releases of sterile mosquitoes with different strategies, J. Biol. Dynam., 9 (2015), 1–14. |
| [14] |
J. Li, J. Li, New revised simple models for interactive wild and sterile mosquito populations and their dynamics, J. Biol. Dynam., 11 (2017), 316–333. doi: 10.1080/17513758.2016.1216613
|
| [15] |
J. Li, L. Cai, Y. Li, Stage-structured wild and sterile mosquito population models and their dynamics, J. Biol. Dynam., 11 (2017), 79–101. doi: 10.1080/17513758.2016.1159740
|
| [16] |
L. Cai, S. Ai, J. Li, Dynamics of mosquitoes populations with different strategies for releasing sterile mosquitoes, SIAM, J. Appl. Math., 74 (2014), 1786–1809. doi: 10.1137/13094102X
|
| [17] |
Y. Dumont, J. M. Tchuenche, Mathematical studies on the sterile insect technique for the Chikungunya disease andAedes albopictus, J. Math. Biol., 65 (2012), 809–854. doi: 10.1007/s00285-011-0477-6
|
| [18] |
J. Huang, S. Ruan, P. Yu, Y. Zhang, Bifurcation analysis of a mosquito population model with a saturated release rate of sterile mosquitoes, SIAM J. Appl. Dyn. Syst., 18 (2019), 939–972. doi: 10.1137/18M1208435
|
| [19] | L. Cai, J. Huang, X. Song, Y. Zhang, Bifurcation analysis of a mosquito population model for proportional releasing sterile mosquitoes, Discrete Contin. Dynam. Syst. Ser. B, 25 (2019), 6279–6295. |
| [20] |
Z. Qiu, X. Wei, C. Shan, H. Zhu, Monotone dynamics and global behaviors of a West Nile virusmodel with mosquito demographics, J. Math. Biol., 80 (2020), 809–834. doi: 10.1007/s00285-019-01442-4
|
| [21] |
M. Huang, X. Song, J. Li, Modelling and analysis of impulsive release of sterile mosquitoes, J. Biol. Dynam., 11 (2017), 147–171. doi: 10.1080/17513758.2016.1254286
|
| [22] |
P. A. Bliman, D. Cardona-Salgado, Y. Dumont, O. Vasilieva, Implementation of control strategies for sterile insect techniques, Math. Biosci., 314 (2019), 43–60. doi: 10.1016/j.mbs.2019.06.002
|
| [23] |
J. Yu, Modeling mosquito population suppression based on delay differential equations, SIAM, J. Appl. Math., 78 (2018), 3168–3187. doi: 10.1137/18M1204917
|
| [24] |
J. Yu, J. Li, Global asymptotic stability in an interactive wild and sterile mosquito model, J. Differ. Equations, 269 (2020), 6193–6215. doi: 10.1016/j.jde.2020.04.036
|
| [25] |
J. Yu, Existence and stability of a unique and exact two periodic orbits for an interactive wild and sterile mosquito model, J. Differ. Equations, 269 (2020), 10395–10415. doi: 10.1016/j.jde.2020.07.019
|
| [26] |
J. Yu, J. Li, Dynamics of interactive wild and sterile mosquitoes with time delay, J. Biol. Dynam., 13 (2019), 606–620. doi: 10.1080/17513758.2019.1682201
|
| [27] | J. Li, S. Ai, Impulsive releases of sterile mosquitoes and interactive dynamics with time delay, J. Biol. Dynam., 14 (2020), 313–331. |
| [28] |
J. Yu, B. Zheng, Modeling Wolbachia infection in mosquito population via discrete dynamical models, J. Differ. Equations Appl., 25 (2019), 1–19. doi: 10.1080/10236198.2018.1551379
|
| [29] |
M. Huang, M. Tang, J. Yu, B. Zheng, A stage structured model of delay differential equations for aedes mosquito population suppression, Discrete Contin. Dynam. Syst., 40 (2020), 3467–3484. doi: 10.3934/dcds.2020042
|
| [30] |
S. Xiang, Y. Pei, X. Liang, Analysis and optimization-based on a sex pheromone and pesticide pest model with gestation delay, Int. J. Biomath., 12 (2019), 1950054. doi: 10.1142/S1793524519500542
|
| [31] |
Z. Q. Yang, X. Y. Wang, Y. N. Zhang, S. B. Vinson, Recent advances in biological control of important native and invasive forest pests in China, Biol. Control, 68 (2014), 117–128. doi: 10.1016/j.biocontrol.2013.06.010
|
| [32] |
P. Neuenschwander, H. R. Herren, Biological control of the cassava mealybug, Phenacoccusmanihoti, by the exotic parasitoid epidinocarsis lopezi in Africa, Philos. Trans. R. Soc. Lond. B, Biol. Sci., 318 (1988), 319–333. doi: 10.1098/rstb.1988.0012
|
| [33] | X. Y. Liang, Y. Z. Pei, M. X. Zhu, Y. F. Lv, Multiple kinds of optimal impulse control strategies on plant-pest-predator model with eco-epidemiology, Appl. Math. Comput., 287 (2016), 1–11. |
| [34] |
Y. Z. Pei, M. M. Chen, X. Y. Liang, C. G. Li, M. X. Zhu, Optimizing pulse timings and amounts of biological interventions for a pest regulation model, Nonlinear Anal. Hybrid Syst., 27 (2018), 353–365. doi: 10.1016/j.nahs.2017.10.003
|
| [35] |
B. Dennis, Allee effects: Population growth, critical density, and the chance of extinction, Nat. Resour. Model., 3 (1989), 481–538. doi: 10.1111/j.1939-7445.1989.tb00119.x
|
| [36] |
S. J. Schreiber, Allee effects, extinctions, and chaotic transients in simple population models, Theor. Popul. Biol., 64 (2003), 201–209. doi: 10.1016/S0040-5809(03)00072-8
|
| [37] | D. D. Bainov, P. S. Simeonov, Impulsive Differential Equations: Periodic Solutions and Applications, CRC Press, London, 66 (1993). |
| [38] | D. D. Bainov, P. S. Simeonov, System with Impulse Effect, Theory and Applications, Prentice Hall, Englewood, (1989). |
| [39] |
H. L. Smith, Monotone dynamical systems: an introduction to the theory of competitive and cooperative systems, Bull. Am. Math. Soc., 33 (1996), 203–209. doi: 10.1090/S0273-0979-96-00642-8
|
| [40] |
K. L. Teo, Control parametrization enhancing transform to optimal control problems, Nonlinear Anal.: Theory, Methods, Appl., 63 (2005), e2223–e2236. doi: 10.1016/j.na.2005.03.066
|
| [41] |
Y. Liu, K. L. Teo, L. S. Jennings, S. Wang, On a class of optimal control problems with state jumps, J. Optim. Theory Appl., 98 (1998), 65–82. doi: 10.1023/A:1022684730236
|
| [42] | F. D. Parker, Management of pest populations by manipulating densities of both hosts and parasites through periodic releases, in Biological Control, Springer, Boston, (1971), 365–376. |
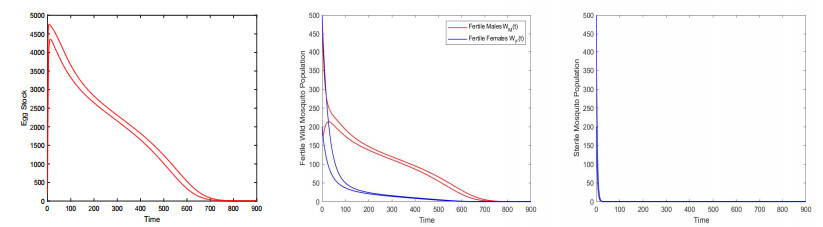
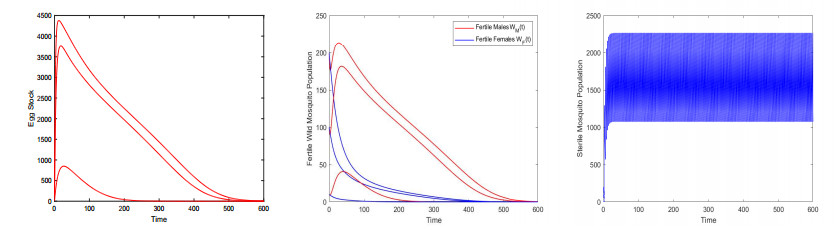
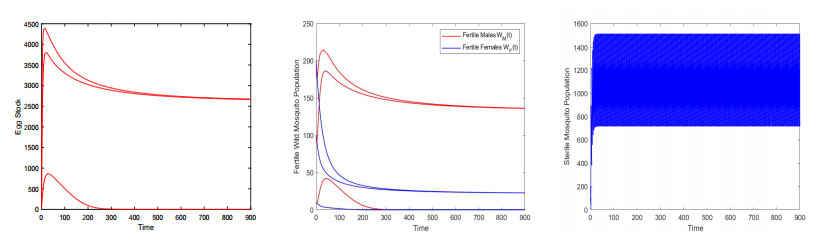
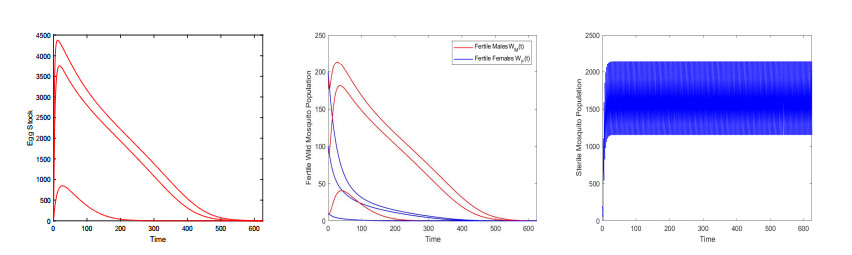
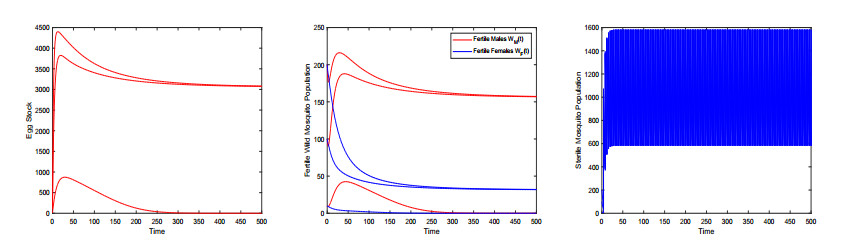
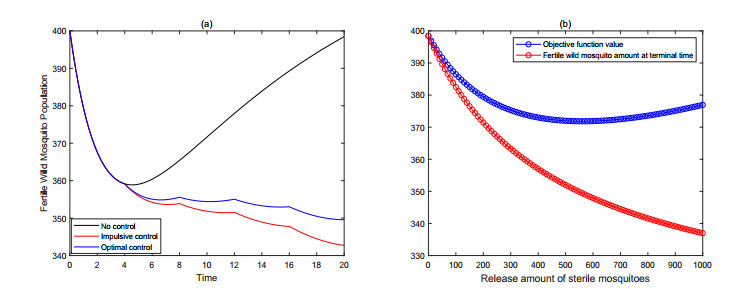
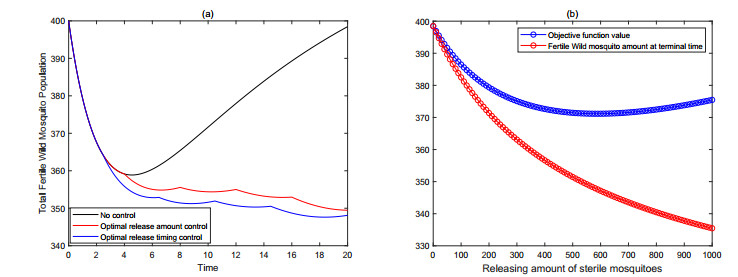
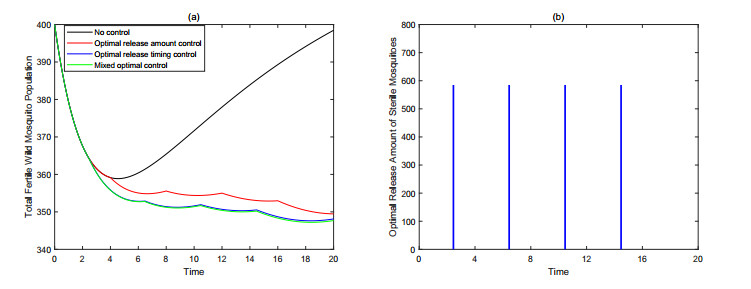
Figures(9) / Tables(3)

Mingzhan Huang, Shouzong Liu, Xinyu Song. Study of the sterile insect release technique for a two-sex mosquito population model[J]. Mathematical Biosciences and Engineering, 2021, 18(2): 1314-1339. doi: 10.3934/mbe.2021069







 DownLoad:
DownLoad: