Citation: Ming-Chun Lai, Jiang-Shan Lian, Wen-Jin Zhang, Jun Xu, Lin Zhou, Shu-Sen Zheng. Compare with safety and efficacy of entecavir and adefovir dipivoxil combination therapy and tenofovir disoproxil fumarate monotherapy for chronic hepatitis B patient with adefovir-resistant[J]. Mathematical Biosciences and Engineering, 2020, 17(1): 627-635. doi: 10.3934/mbe.2020032

| [1] | J. J. Ott, G. A. Stevens, J. Groeger, et al., Global epidemiology of hepatitis B virus infection: New estimates of age-specific HBsAg seroprevalence and endemicity, Vaccine, 30 (2012); 2212-2219. |
| [2] | U. H. Iloeje, H. I. Yang, J. Su, et al., Predicting cirrhosis risk based on the level of circulating hepatitis B viral load, Gastroenterology, 130 (2006), 678-686. |
| [3] | G. A. Kim, Y. S. Lim, S. Han, et al., High risk of hepatocellular carcinoma and death in patients with immune-tolerant-phase chronic hepatitis B, Gut, 67 (2018), 945-952. |
| [4] | M. Aizawa, A. Tsubota, K. Fujise, et al., Clinical course and predictive factors of virological response in long-term lamivudine plus adefovir dipivoxil combination therapy for lamivudine-resistant chronic hepatitis B patients, J. Med. Virol., 83 (2011), 953-961. |
| [5] |
R. P. Perrillo, H. W. Hann, E. Schiff, et al., Extended treatment with lamivudine and adefovir dipivoxil in chronic hepatitis B patients with lamivudine resistance, Hepatol. Int., 5 (2011), 654-663. doi: 10.1007/s12072-010-9228-9

|
| [6] | N. A. Terrault, N. H. Bzowej, K. M. Chang, et al., AASLD guidelines for treatment of chronic hepatitis B, Hepatology, 63 (2016), 261-283. |
| [7] | C. L. Lai, J. Dienstag, E. Schiff, et al., Prevalence and clinical correlates of YMDD variants during lamivudine therapy for patients with chronic hepatitis B, Clin. Infect. Dis., 36 (2003), 687-696. |
| [8] | W. Ke, L. Liu, C. Zhang, et al., Comparison of efficacy and safety of tenofovir and entecavir in chronic hepatitis B virus infection: A systematic review and meta-analysis, PLoS One, 9 (2014), e98865. |
| [9] | F. Güzelbulut, A. O. Ovünç, Z. A. Oetinkaya, et al., Comparison of the efficacy of entecavir and tenofovir in chronic hepatitis B, Hepatogastroenterology, 59 (2012), 477-480. |
| [10] | Z. B. Huang, S. S. Zhao, Y. Huang, et al., Comparison of the Efficacy of Lamivudine Plus Adefovir Versus Entecavir in the Treatment of Lamivudine-Resistant Chronic Hepatitis B: A Systematic Review and Meta-Analysis, Clin. Ther., 35 (2013), 1997-2006. |
| [11] | A. S. Lok, B. J. McMahon, R. S. Brown, et al., Antiviral therapy for chronic hepatitis B viral infection in adults: A systematic review and meta-analysis, Hepatology, 63 (2016), 284-306. |
| [12] | W. K. Seto, Y. F. Lam, J. Fung, et al., Changes of HBsAg and HBV DNA levels in Chinese chronic hepatitis B patients after 5 years of entecavir treatment, J. Gastroenterol. Hepatol., 29 (2014), 1028-1034. |
| [13] | M. Buti, N. Tsai, J. Petersen, et al., Seven-year efficacy and safety of treatment with tenofovir disoproxil fumarate for chronic hepatitis B virus infection, Dig. Dis. Sci., 60 (2015), 1457-1464. |
| [14] |
Y. F. Liaw, I. S. Sheen, C. M. Lee, et al., Tenofovir disoproxil fumarate (TDF), emtricitabine/TDF, and entecavir in patients with decompensated chronic hepatitis B liver disease, Hepatology, 53 (2011), 62-72. doi: 10.1002/hep.23952
|
| [15] | T. G. Vassiliadis, O. Giouleme, G. Koumerkeridis, et al., Adefovir plus lamivudine are more effective than adefovir alone in lamivudine-resistant HBeAg- chronic hepatitis B patients: A 4-year study, J. Gastroenterol. Hepatol., 25 (2010), 54-60. |
| [16] | M. Tanaka, F. Suzuki, Y. Seko, et al., Renal dysfunction and hypophosphatemia during long-term lamivudine plus adefovir dipivoxil therapy in patients with chronic hepatitis B, J. Gastroenterol., 49 (2014), 470-480. |
| [17] | M. L. Chang and Y. F. Liaw, Hepatitis B flares in chronic hepatitis B: Pathogenesis, natural course, and management, J. Hepatol., 61 (2014), 1407-1417. |
| [18] | P. Thurairajah, A. Khanna and D. Mutimer, Add-on combination therapy with adefovir dipivoxil induces renal impairment in patients with lamivudine-refractory hepatitis B virus (J. Viral. Hepat. 2010 Feb 1;17(2):123-9), J. Viral. Hepat., 18 (2011), 810-820. |
| [19] | Y. J. Kim, H. C. Cho, D. H. Sinn, et al., Frequency and risk factors of renal impairment during long-term adefovir dipivoxil treatment in chronic hepatitis B patients, J. Gastroenterol. Hepatol., 27 (2012), 306-312. |
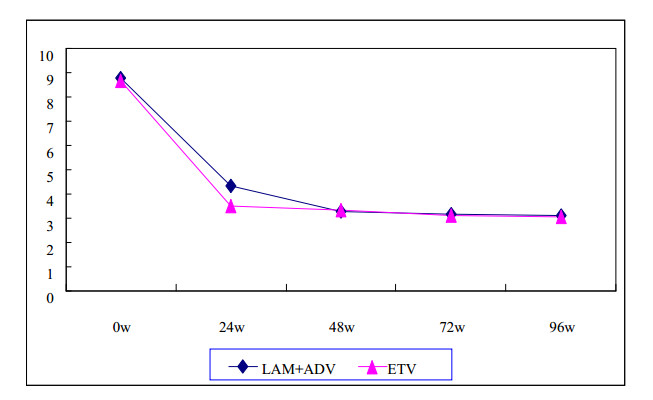
Figures(1) / Tables(2)

Ming-Chun Lai, Jiang-Shan Lian, Wen-Jin Zhang, Jun Xu, Lin Zhou, Shu-Sen Zheng. Compare with safety and efficacy of entecavir and adefovir dipivoxil combination therapy and tenofovir disoproxil fumarate monotherapy for chronic hepatitis B patient with adefovir-resistant[J]. Mathematical Biosciences and Engineering, 2020, 17(1): 627-635. doi: 10.3934/mbe.2020032







 DownLoad:
DownLoad: