Citation: Yonghua Zhuang, Kristen Wade, Laura M. Saba, Katerina Kechris. Development of a tissue augmented Bayesian model for expression quantitative trait loci analysis[J]. Mathematical Biosciences and Engineering, 2020, 17(1): 122-143. doi: 10.3934/mbe.2020007

| [1] | A. C. Nica and E. T. Dermitzakis, Expression quantitative trait loci: present and future, Philos. Trans. R. Soc. Lond. B Biol. Sci., 368 (2013), 20120362. |
| [2] | L. A. Hindorff, P. Sethupathy, H. A. Junkins, et al., Potential etiologic and functional implications of genome-wide association loci for human diseases and traits, Proc. Natl. Acad. Sci. U S A, 106 (2009), 9362-9367. |
| [3] |
B. Hrdlickova, R. C. de Almeida, Z. Borek, et al., Genetic variation in the non-coding genome: Involvement of micro-rnas and long non-coding rnas in disease, Biochim. Biophys. Acta, 1842 (2014), 1910-1922. doi: 10.1016/j.bbadis.2014.03.011

|
| [4] | I. Ricaño-Ponce and C. Wijmenga, Mapping of immune-mediated disease genes, Annu. Rev. Genomics Hum. Genet., 14 (2013), 325-353. |
| [5] | R. C. Jansen and J. P. Nap, Genetical genomics: the added value from segregation, Trends Genet., 17 (2001), 388-391. |
| [6] | L. J. Carithers, K. Ardlie, M. Barcus, et al., A novel approach to high-quality postmortem tissue procurement: The gtex project, Biopreserv. Biobank., 13 (2015), 311-319, |
| [7] | W. Cookson, L. Liang, G. Abecasis, et al., Mapping complex disease traits with global gene expression, Nat. Rev. Genet., 10 (2009), 184-194. |
| [8] | A. C. Nica and E. T. Dermitzakis, Using gene expression to investigate the genetic basis of complex disorders, Hum. Mol. Genet., 17 (2008), R129-134. |
| [9] | M. V. Rockman and L. Kruglyak, Genetics of global gene expression, Nat. Rev. Genet., 7 (2006), 862-872. |
| [10] | F. A. Cubillos, V. Coustham and O. Loudet, Lessons from eqtl mapping studies: non-coding regions and their role behind natural phenotypic variation in plants, Curr. Opin. Plant. Biol., 15 (2012), 192-198. |
| [11] | H. B. Fraser, A. M. Moses and E. E. Schadt, Evidence for widespread adaptive evolution of gene expression in budding yeast, Proc. Natl. Acad. Sci. U S A, 107 (2010), 2977-2982. |
| [12] | A. L. Dixon, L. Liang, M. F. Moffatt, et al., A genome-wide association study of global gene expression, Nat. Genet., 39 (2007), 1202-1207. |
| [13] | H. H. H. Göring, J. E. Curran, M. P. Johnson, et al., Discovery of expression qtls using large-scale transcriptional profiling in human lymphocytes, Nat. Genet., 39 (2007), 1208-1216. |
| [14] | E. E. Schadt, C. Molony, E. Chudin, et al., Mapping the genetic architecture of gene expression in human liver, PLoS Biol., 6 (2008), e107. |
| [15] |
A. Gerrits, Y. Li, B. M. Tesson, et al., Expression quantitative trait loci are highly sensitive to cellular differentiation state, PLoS Genet., 5 (2009), e1000692. doi: 10.1371/journal.pgen.1000692
|
| [16] | G. K. Chen and J. S. Witte, Enriching the analysis of genomewide association studies with hierarchical modeling, Am. J. Hum. Genet., 81 (2007), 397-404. |
| [17] | X. Zhang, S. Huang, W. Sun, et al., Rapid and robust resampling-based multiple-testing correction with application in a genome-wide expression quantitative trait loci study, Genetics, 190 (2012), 1511-1520. |
| [18] | M. P. Scott-Boyer, G. C. Imholte, A. Tayeb, et al., An integrated hierarchical bayesian model for multivariate eqtl mapping, Stat. Appl. Genet. Mol. Biol., 11 (2012), 10.1515/1544-6115.1760. |
| [19] | O. Stegle, L. Parts, R. Durbin, et al., A bayesian framework to account for complex non-genetic factors in gene expression levels greatly increases power in eqtl studies, PLoS Comput. Biol., 6 (2010), e1000770. |
| [20] | M. Stephens and D. J. Balding, Bayesian statistical methods for genetic association studies, Nat. Rev. Genet., 10 (2009), 681-690. |
| [21] | J.-B. Veyrieras, S. Kudaravalli, S. Y. Kim, et al., High-resolution mapping of expression-qtls yields insight into human gene regulation, PLoS Genet., 4 (2008), e1000214. |
| [22] | M. Banterle, L. Bottolo, S. Richardson, et al., Sparse variable and covariance selection for highdimensional seemingly unrelated bayesian regression, bioRxiv, 467019. |
| [23] | G. C. Imholte, M.-P. Scott-Boyer, A. Labbe, et al., ibmq: a r/bioconductor package for integrated bayesian modeling of eqtl data, Bioinformatics, 29 (2013), 2797-2798. |
| [24] | D. Duong, L. Gai, S. Snir, et al., Applying meta-analysis to genotype-tissue expression data from multiple tissues to identify eqtls and increase the number of egenes, Bioinformatics, 33 (2017), i67-i74. |
| [25] | J. H. Sul, B. Han, C. Ye, et al., Effectively identifying eqtls from multiple tissues by combining mixed model and meta-analytic approaches, PLoS Genet., 9 (2013), e1003491. |
| [26] | T. Flutre, X. Wen, J. Pritchard, et al., A statistical framework for joint eqtl analysis in multiple tissues, PLoS Genet., 9 (2013), e1003486. |
| [27] | G. Li, A. A. Shabalin, I. Rusyn, et al., An empirical bayes approach for multiple tissue eqtl analysis, Biostatistics, 19 (2018), 391-406. |
| [28] | A. Das, M. Morley, C. S. Moravec, et al., Bayesian integration of genetics and epigenetics detects causal regulatory snps underlying expression variability, Nat. Commun., 6 (2015), 8555. |
| [29] | E. J. Chesler, L. Lu, J. Wang, et al., Webqtl: rapid exploratory analysis of gene expression and genetic networks for brain and behavior, Nat. Neurosci., 7 (2004), 485-486. |
| [30] | J. Wang, R. W. Williams and K. F. Manly, Webqtl: web-based complex trait analysis, Neuroinformatics, 1 (2003), 299-308. |
| [31] |
T. J. Phillips, M. Huson, C. Gwiazdon, et al., Effects of acute and repeated ethanol exposures on the locomotor activity of bxd recombinant inbred mice, Alcohol. Clin. Exp. Res., 19 (1995), 269-278. doi: 10.1111/j.1530-0277.1995.tb01502.x
|
| [32] |
B. Tabakoff, L. Saba, K. Kechris, et al., The genomic determinants of alcohol preference in mice, Mamm. Genome., 19 (2008), 352-365. doi: 10.1007/s00335-008-9115-z
|
| [33] |
B. J. Bennett, C. R. Farber, L. Orozco, et al., A high-resolution association mapping panel for the dissection of complex traits in mice, Genome. Res., 20 (2010), 281-290. doi: 10.1101/gr.099234.109
|
| [34] | R. Alberts, L. Lu, R. W. Williams, et al., Genome-wide analysis of the mouse lung transcriptome reveals novel molecular gene interaction networks and cell-specific expression signatures, Respir. Res., 12 (2011), 61. |
| [35] |
C. Blauwendraat, M. Francescatto, J. R. Gibbs, et al., Comprehensive promoter level expression quantitative trait loci analysis of the human frontal lobe, Genome Med., 8 (2016), 65. doi: 10.1186/s13073-016-0320-1
|
| [36] |
J. A. Webster, J. R. Gibbs, J. Clarke, et al., Genetic control of human brain transcript expression in alzheimer disease, Am. J. Hum. Genet., 84 (2009), 445-458. doi: 10.1016/j.ajhg.2009.03.011
|
| [37] |
S. Lagarrigue, L. Martin, F. Hormozdiari, et al., Analysis of allele-specific expression in mouse liver by rna-seq: a comparison with cis-eqtl identified using genetic linkage, Genetics, 195 (2013), 1157-1166. doi: 10.1534/genetics.113.153882
|
| [38] | A. Gelman, J. B. Carlin, H. S. Stern, et al., Bayesian data analysis, vol. 2, Chapman & Hall/CRC Boca Raton, FL, USA, 2014. |
| [39] | P. D. Hoff, A first course in Bayesian statistical methods, vol. 580, Springer, 2009. |
| [40] | E. Lesaffre and A. B. Lawson, Bayesian biostatistics, John Wiley & Sons, 2012. |
| [41] | X. Robin, N. Turck, A. Hainard, et al., proc: an open-source package for r and s+ to analyze and compare roc curves, BMC Bioinformatics, 12 (2011), 77. |
| [42] | E. R. DeLong, D. M. DeLong and D. L. Clarke-Pearson, Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach, Biometrics, 44 (1988), 837-845. |
| [43] | S. A. Stouffer, E. A. Suchman, L. C. DeVinney, et al., The american soldier: Adjustment during army life, Princeton University Press, Vol. 1. |
| [44] | L. T., On the combination of independent tests, Magyar Tud Akad Mat Kutato Int Közl. |
| [45] | M. C. Whitlock, Combining probability from independent tests: the weighted z-method is superior to fisher's approach, J. Evol. Biol., 18 (2005), 1368-1373. |
| [46] | D. Bates, M. Mächler, B. Bolker, et al., Fitting linear mixed-effects models using lme4, J. Stat. Software, 67 (2015), 1-48. |
| [47] | R. V. Lenth, Least-squares means: The R package lsmeans, J. Stat. Software, 69 (2016), 1-33. |
| [48] | RStudio Team, RStudio: Integrated Development Environment for R, RStudio, Inc., Boston, MA, 2015. |
| [49] | R Core Team, R: A Language and Environment for Statistical Computing, R Foundation for Statistical Computing, Vienna, Austria, 2015. |
| [50] | A. A. Shabalin, Matrix eqtl: ultra fast eqtl analysis via large matrix operations, Bioinformatics, 28 (2012), 1353-1358. |
| [51] | H. Wickham, ggplot2: Elegant Graphics for Data Analysis, Springer-Verlag New York, 2009. |
| [52] | R. C. Team, D. Wuertz, T. Setz, et al., fBasics: Rmetrics-Markets and Basic Statistics, 2014, R package version 3011.87. |
| [53] | D. B. Dahl, xtable: Export Tables to LaTeX or HTML, 2016, R package version 1.8-2. |
| [54] | S. Durinck, Y. Moreau, A. Kasprzyk, et al., Biomart and bioconductor: a powerful link between biological databases and microarray data analysis, Bioinformatics, 21 (2005), 3439-3440. |
| [55] | H. Wickham, The split-apply-combine strategy for data analysis, J. Stat. Software, 40 (2011), 1-29. |
| [56] | M. Dowle, A. Srinivasan, T. Short, et al., data.table: Extension of Data.frame, 2015, R package version 1.9.6. |
| [57] | S. Anders and W. Huber, Differential expression analysis for sequence count data, Genome biol., 11 (2010), R106. |
| [58] | C. W. Law, Y. Chen, W. Shi, et al., voom: Precision weights unlock linear model analysis tools for rna-seq read counts, Genome Biol., 15 (2014), R29. |
| [59] | W. Sun, A statistical framework for eqtl mapping using rna-seq data, Biometrics, 68 (2012), 1-11. |
| [60] | L. Bottolo and S. Richardson, Evolutionary stochastic search for bayesian model exploration, Bayesian Anal., 5 (2010), 583-618. |
| [61] | N. A. Walter, S. K. McWeeney, S. T. Peters, et al., Snps matter: impact on detection of differential expression, Nature Methods, 4 (2007), 679. |
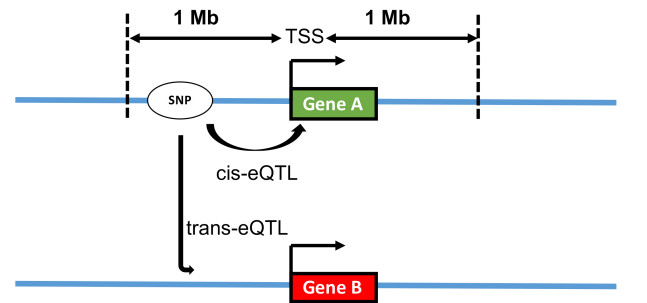
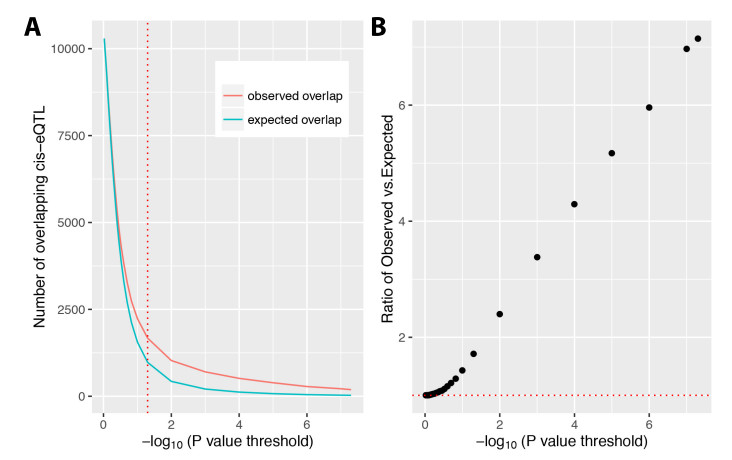
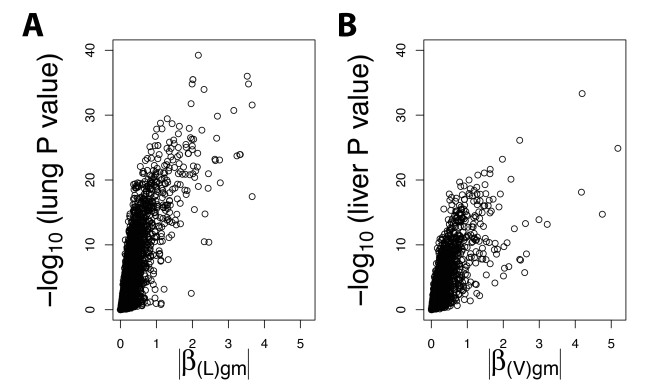
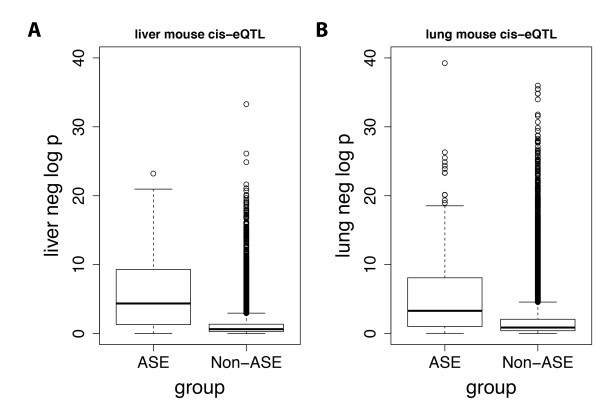
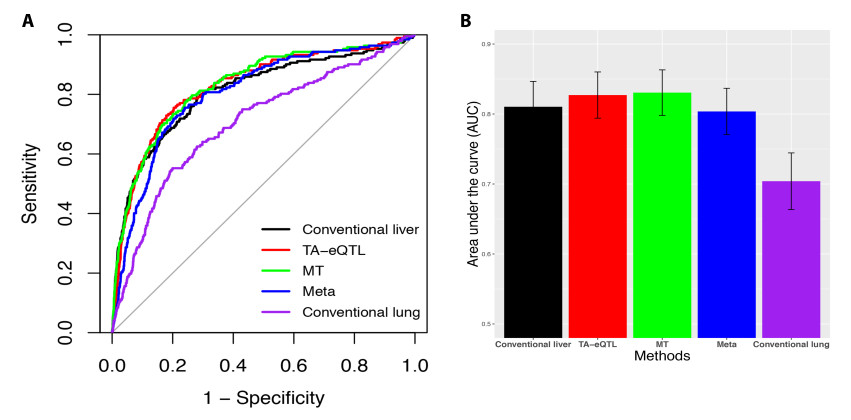
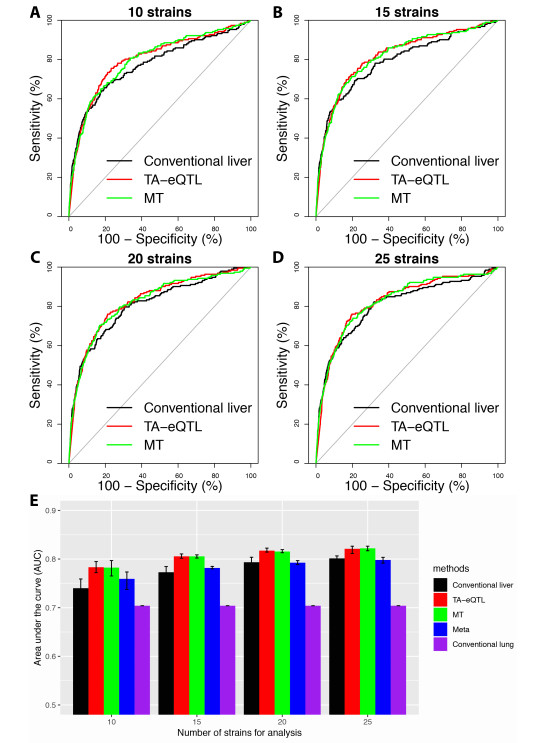
Figures(6) / Tables(2)

Yonghua Zhuang, Kristen Wade, Laura M. Saba, Katerina Kechris. Development of a tissue augmented Bayesian model for expression quantitative trait loci analysis[J]. Mathematical Biosciences and Engineering, 2020, 17(1): 122-143. doi: 10.3934/mbe.2020007







 DownLoad:
DownLoad: