Citation: Tuoi Vo, William Lee, Adam Peddle, Martin Meere. Modelling chemistry and biology after implantation of a drug-eluting stent. Part Ⅰ: Drug transport[J]. Mathematical Biosciences and Engineering, 2017, 14(2): 491-509. doi: 10.3934/mbe.2017030

| [1] | [ J. P. Alberding,A. L. Baldwin,J. K. Barton,E. Wiley, Effects of pulsation frequency and endothelial integrity on enhanced arterial transmural filtration produced by pulsatile pressure, Am. J. Physiol. Heart Circ. Physiol., 289 (2005): H931-H937. |
| [2] | [ A. L. Baldwin,L. M. Wilson,I. Gradus-Pizlo,R. Wilensky,K. March, Effect of atherosclerosis on transmural convection and arterial ultrastructure, JArterioscler. Thromb. Vasc. Biol., 17 (1997): 3365-3375. |
| [3] | [ J. Bennett,C. Dubois, A novel platinum chromium everolimus-eluting stent for the treatment of coronary artery disease, Biologics: Targets and Therapy, 17 (2013): 149-159. |
| [4] | [ P. Biscari,S. Minisini,D. Pierotti,G. Verzini,P. Zunino, Controlled release with finite dissolution rate, SIAM Journal on Applied Mathematics, 71 (2011): 731-752. |
| [5] | [ A. Borghi,E. Foa,R. Balossino,F. Migliavacca,G. Dubini, Modelling drug elution from stents: Effects of reversible binding in the vascular wall and degradable polymeric matrix, Computer Methods in Biomechanics and Biomedical Engineering, 11 (2008): 367-377. |
| [6] | [ F. Bozsak,J. Chomaz,A. I. Barakat, Modeling the transport of drugs eluted from stents: Physical phenomena driving drug distribution in the arterial wall, Biomech Model Mechanobiol, 13 (2014): 327-347. |
| [7] | [ D. Capodanno,F. Dipasqua,C. Tamburino, Novel drug-eluting stents in the treatment of de novo coronary lesions, Vasc Health Risk Management, 7 (2011): 103-118. |
| [8] | [ D. S. Cohen,T. Erneux, Controlled drug release asymptotics, SIAM Journal on Applied Mathematics, 58 (1998): 1193-1204. |
| [9] | [ C. Conway,J. P. McGarry,P. E. McHugh, Modelling of atherosclerotic plaque for use in a computational test-bed for stent angioplasty, Annals of Biomedical Engineering, 42 (2014): 2425-2439. |
| [10] | [ G. Frenning, Theoretical analysis of the release of slowly dissolving drugs from spherical matrix systems, Journal of Controlled Release, 95 (2004): 109-117. |
| [11] | [ M. J. Lever,J. M. Tarbell,C. G. Caro, The effect of luminal flow in rabbit carotid artery on transmural fluid transport, Experimental Physiology, 77 (1992): 553-563. |
| [12] | [ A. D. Levin,N. Vukmirovic,C. Hwang,E. R. Edelman, Specific binding to intracellular proteins determines arterial transport properties for rapamycin and paclitaxel, PNAS, 101 (2004): 9463-9467. |
| [13] | [ M. A. Lovich,E. R. Edelman, Computational simulations of local vascular heparin deposition and distribution, American Journal of Physiology, 271 (1996): H2014-H2024. |
| [14] | [ D. M. Martin,F. J. Boyle, Drug-eluting stents for coronary artery disease: A review, Medical Engineering & Physics, 33 (2011): 148-163. |
| [15] | [ S. McGinty, A decade of modelling drug release from arterial stents, Mathematical Bioscience, 257 (2014): 80-90. |
| [16] | [ S. McGinty,S. McKee,C. McCormick,M. Wheel, Release mechanism and parameter estimation in drug-eluting stent systems: Analytical solutions of drug release and tissue transport, Mathematical Medicine and Bilology, 32 (2015): 163-186. |
| [17] | [ S. McGinty,S. McKee,R. M. Wadsworth,C. McCormick, Modelling drug-eluting stents, Mathematical Medicine and Bilology, 28 (2011): 1-29. |
| [18] | [ S. McGinty,G. Pontrelli, A general model of coupled drug release and tissue absorption for drug delivery devices, Journal of Controlled Release, 217 (2015): 327-336. |
| [19] | [ A. Peddle, T. T. N. Vo and W. Lee, Modelling chemistry and biology after implantation of a drug-eluting stent. Part Ⅱ: Cell proliferation, in progress. |
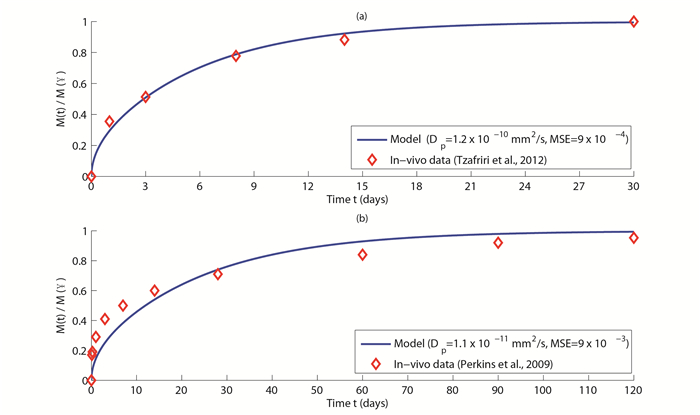
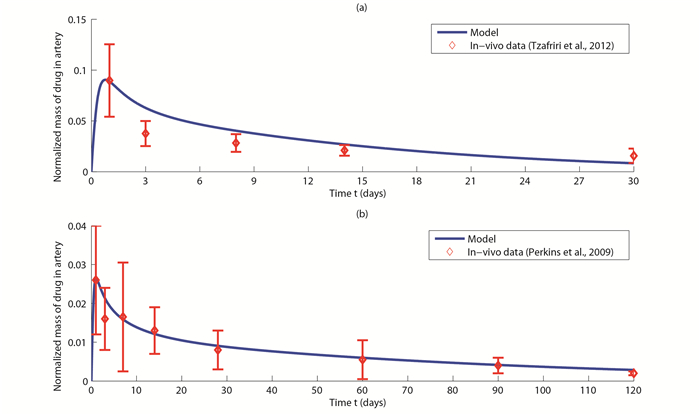
| [20] | [ L. E. L. Perkins,K. H. Boeke-Purkis,Q. Wang,S. K. Stringer,L. A. Coleman, XIENCE V Everolimus-eluting coronary stent system: A preclinical assessment, Journal of Interventional Cardiology, 22 (2009): S28-S40. |
| [21] | [ D. V. Sakharov,L. V. Kalachev,D. C. Rijken, Numerical simulation of local pharmacokinetics of a drug after intravascular delivery with an eluting stent, Journal of Drug Targeting, 10 (2002): 507-513. |
| [22] | [ R. W. Sirianni,E. Jang,K. M. Miller,W. M. Saltzman, Parameter estimation methodology in a model of hydrophobic drug release from a polymer coating, Journal of Controlled Release, 142 (2010): 474-482. |
| [23] | [ A. Tedgui,M. J. Lever, Filtration through damaged and undamaged rabbit thoracic aorta, Am. J. Physiol., 247 (1984): H784-H791. |
| [24] | [ A. R. Tzafriri,A. Groothuis,G. S. Price,E. R. Edelman, Stent elution rate determines drug deposition and receptor-mediated effects, Journal of Controlled Release, 161 (2012): 918-926. |
| [25] | [ A. R. Tzafriri,A. D. Levin,E. R. Edelman, Diffusion-limited binding explains binary dose response for local arterial and tumor drug delivery, Cell Proliferation, 42 (2009): 348-363. |
| [26] | [ T. T. N. Vo,R. Yang,Y. Rochev,M. Meere, A mathematical model for drug delivery, Progress in Industrial Mathematics at ECMI 2010, Mathematics in Industry, 17 (2012): 521-528. |
| [27] | [ T. T. N. Vo, Mathematical Analysis of Some Models for Drug Delivery PhD thesis, National University of Ireland Galway, 2012. |
| [28] | [ P. Zunino, Multidimensional pharmacokinetic models applied to the design of drug-eluting stents, Cardiovascular Engineering, 4 (2004): 181-191. |
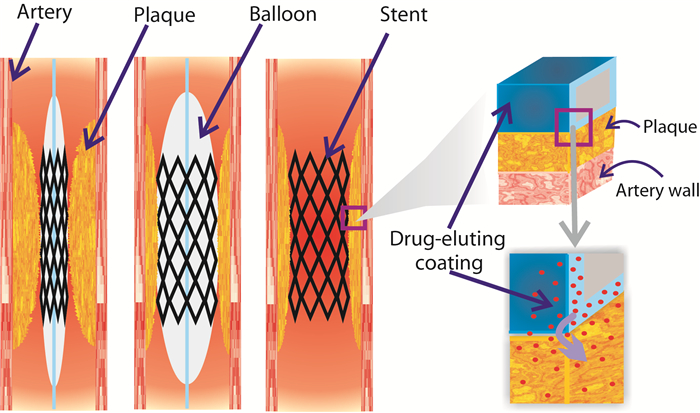
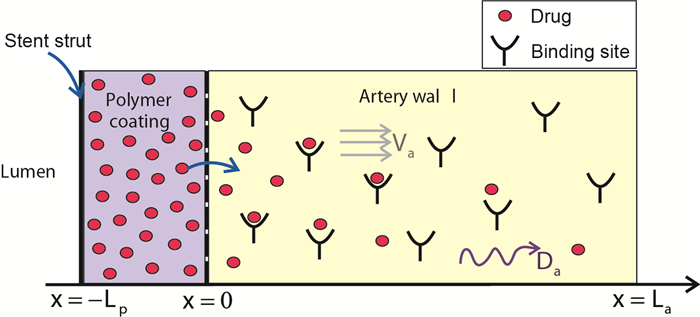
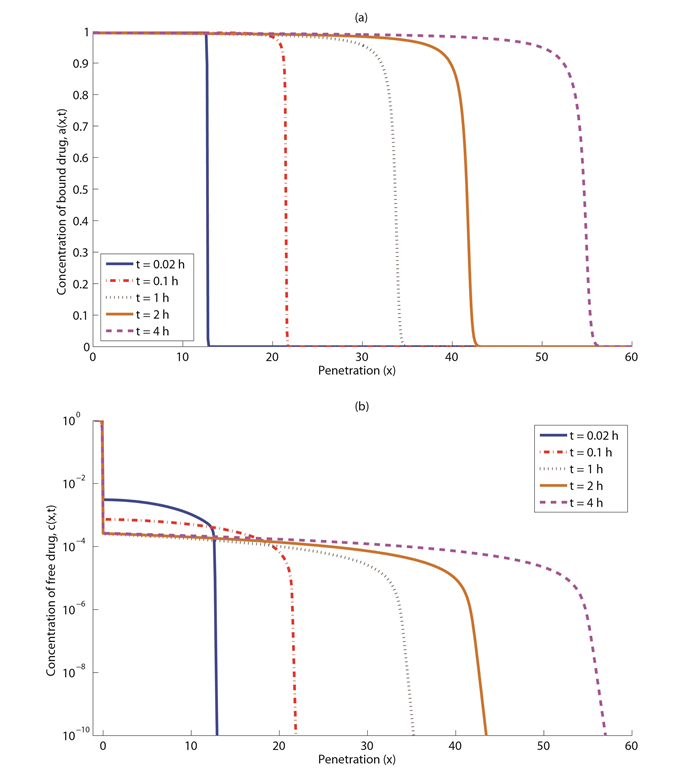
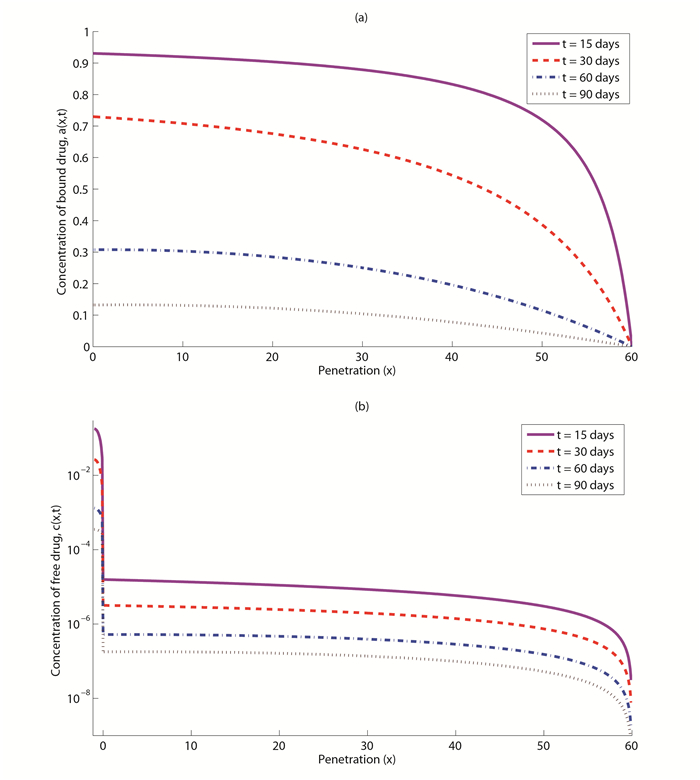
Figures(7) / Tables(5)

Tuoi Vo, William Lee, Adam Peddle, Martin Meere. Modelling chemistry and biology after implantation of a drug-eluting stent. Part Ⅰ: Drug transport[J]. Mathematical Biosciences and Engineering, 2017, 14(2): 491-509. doi: 10.3934/mbe.2017030







 DownLoad:
DownLoad: