In practice, objective condition may impose constraints on design region, which make it difficult to find the exact D-optimal design. In this paper, we propose a Multi-stage Differential Evolution (MDE) algorithm to find the global approximated D-optimal design in an experimental region with linear or nonlinear constraints. MDE algorithm is approved from Differential Evolution (DE) algorithm. It has low requirements for both feasible regions and initial values. In iteration, MDE algorithm pursues evolutionary equilibrium rather than convergence speed, so it can stably converge to the global D-optimal design instead of the local ones. The advantages of MDE algorithm in finding D-optimal design will be illustrated by examples.
Citation: Xinfeng Zhang, Zhibin Zhu, Chongqi Zhang. Multi-stage differential evolution algorithm for constrained D-optimal design[J]. AIMS Mathematics, 2021, 6(3): 2956-2969. doi: 10.3934/math.2021179
In practice, objective condition may impose constraints on design region, which make it difficult to find the exact D-optimal design. In this paper, we propose a Multi-stage Differential Evolution (MDE) algorithm to find the global approximated D-optimal design in an experimental region with linear or nonlinear constraints. MDE algorithm is approved from Differential Evolution (DE) algorithm. It has low requirements for both feasible regions and initial values. In iteration, MDE algorithm pursues evolutionary equilibrium rather than convergence speed, so it can stably converge to the global D-optimal design instead of the local ones. The advantages of MDE algorithm in finding D-optimal design will be illustrated by examples.

| [1] |
D. C. Montgomery, E. N. Loredo, D. Jearkpaporn, M. C. Testik, Experimental designs for constrained regions, Qual. Eng., 14 (2002), 587–601. doi: 10.1081/QEN-120003561

|
| [2] |
J. J. Borkowski, G. F. Piepel, Uniform designs for highly constrained mixture experiments, J. Qual. Technol., 41 (2009), 35–47. doi: 10.1080/00224065.2009.11917758
|
| [3] |
R. D. Snee, D. W. Marquardt, Extreme vertices designs for linear mixture models, Technometrics, 16 (1974), 399–408. doi: 10.1080/00401706.1974.10489209
|
| [4] | T. J. Mitchell, An Algorithm for the construction of D-optimal experimental designs, Technometrics, 16 (1974), 203–210. |
| [5] |
R. D. Cook, C. J. Nachtsheim, A comparison of algorithms for constructing exact D-optimal designs, Technometrics, 22 (1980), 315–324. doi: 10.1080/00401706.1980.10486162
|
| [6] |
A. Heredia-Langner, D. C. Montgomery, W. M. Carlyle, C. M. Borror, Model-robust optimal designs: A genetic algorithm approach, J. Qual. Technol., 36 (2004), 263–279. doi: 10.1080/00224065.2004.11980273
|
| [7] |
A. Heredia-Langner, W. M. Carlyle, D. C. Montgomery, C. M. Borror, G. C. Runger, Genetic algorithms for the construction of D-optimal designs, J. Qual. Technol., 35 (2003), 28–46. doi: 10.1080/00224065.2003.11980189
|
| [8] | Y. Yu, D-optimal designs via a cocktail algorithm, Stat. Comput., 21 (2010), 475–481. |
| [9] | Y. Liu, M. Q. Liu, Construction of uniform designs for mixture experiments with complex constraints, Commun. Stat.-Theor. M., 45 (2015), 2172–2180. |
| [10] | G. Li, C. Zhang, Random search algorithm for optimal mixture experimental design, Commun. Stat. Theor. M., 47 (2017), 1413–1422. |
| [11] |
J. Duan, W. Gao, H. K. T. Ng, Efficient computational algorithm for optimal continuous experimental designs, J. Comput. Appl. Math., 350 (2019), 98–113. doi: 10.1016/j.cam.2018.09.046
|
| [12] | W. K. Wong, R. B. Chen, C. C. Huang, W. Wang, A modified particle swarm optimization technique for finding optimal designs for mixture models, PLoS One, 10 (2015), e0124720. |
| [13] |
P. Y. Yang, F. I. Chou, J. T. Tsai, J. H. Chou, Adaptive uniform experimental design based fractional order particle swarm optimizer with non-linear time varying evolution, Appl. Sci., 9 (2019), 5537. doi: 10.3390/app9245537
|
| [14] | K. Price, Differential evolution: A fast and simple numerical optimizer, In: Proceedings of North American Fuzzy Information Processing, 1996,524–527. |
| [15] |
R. Storn, K. Price, Differential Evolution — A Simple and Efficient Heuristic for global optimization over continuous spaces, J. Global. Optim., 11 (1997), 341–359. doi: 10.1023/A:1008202821328
|
| [16] | K. V. Price, R. M. Storn, J. A. Lampinen, Differential evolution-A practical approach to global optimization, Springer, Berlin, 2005. |
| [17] |
Z. Zhao, J. Z. Wang, J. Zhao, Z. Y. Su, Using a Grey model optimized by Differential Evolution algorithm to forecast the per capita annual net income of rural households in China, Omega, 40 (2012), 525–532. doi: 10.1016/j.omega.2011.10.003
|
| [18] |
K. Rajesh, A. Bhuvanesh, S. Kannan, C. Thangaraj, Least cost generation expansion planning with solar power plant using Differential Evolution algorithm, Renew. Energ., 85 (2016), 677–686. doi: 10.1016/j.renene.2015.07.026
|
| [19] | W. Xu, W. K. Wong, K. C. Tan, J. Xu, Finding high-dimensional D-optimal designs for logistic models via differential evolution, IEEE Access 7 (2019), 7133–7146. |
| [20] | W. Deng, J. J. Xu, X. Z. Gao, H. M. Zhao, An enhanced MSIQDE algorithm with novel multiple strategies for global optimization problems, IEEE T. Syst. Man Cy-S., (2020), 1–10. |
| [21] | J. Kiefer, Optimum experimental designs, J. R. Stat. Soc. B., 21 (1959), 272–319. |
| [22] | R. H. Myers, D. C. Montgomery, G. G. Vining, T. J. Robinson, Generalized Linear Models: With Applications in Engineering and the Sciences, 2 Eds., John Wiely & Sons, New York, 2002. |
| [23] |
A. Dasgupta, S. Mukhopadhyay, W. J. Studden, Compromise designs in heteroscedastic linear models, J. Stat. Plan. Infer., 32 (1992), 363–384. doi: 10.1016/0378-3758(92)90017-M
|
| [24] | K. Chaloner, Optimal bayesian experimental design for linear models, Ann. Stat., 12 (1984), 283–300. |
| [25] |
R. D. Cook, V. V. Fedorov, Constrained optimization of experimental design, Statistics, 26 (1995), 129–178. doi: 10.1080/02331889508802474
|
| [26] | J. A. Cornell, Experiments with mixtures: designs, models and the alysis of mixture data, 3 Eds., John Wiely & Sons, New York, 2002. |
| [27] | J. Kiefer, J. Wolfowitz, Optimum extrapolation and interpolation designs Ⅱ, Ann. I. Stat. Math. 16 (1964), 295–303. |
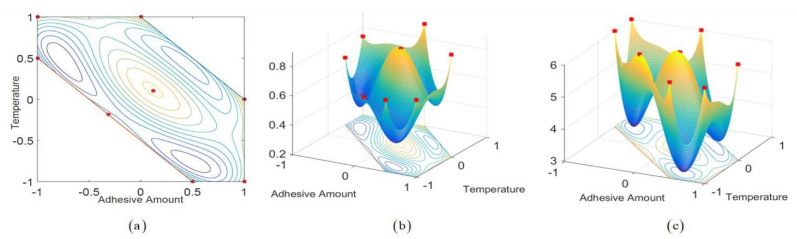
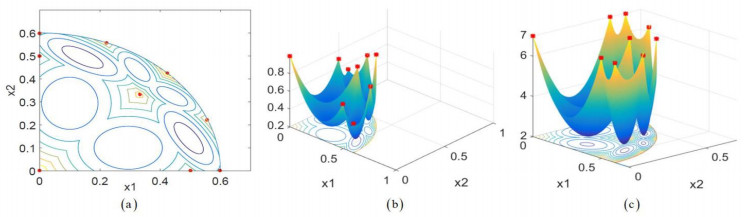
Figures(2) / Tables(4)

Xinfeng Zhang, Zhibin Zhu, Chongqi Zhang. Multi-stage differential evolution algorithm for constrained D-optimal design[J]. AIMS Mathematics, 2021, 6(3): 2956-2969. doi: 10.3934/math.2021179







 DownLoad:
DownLoad: