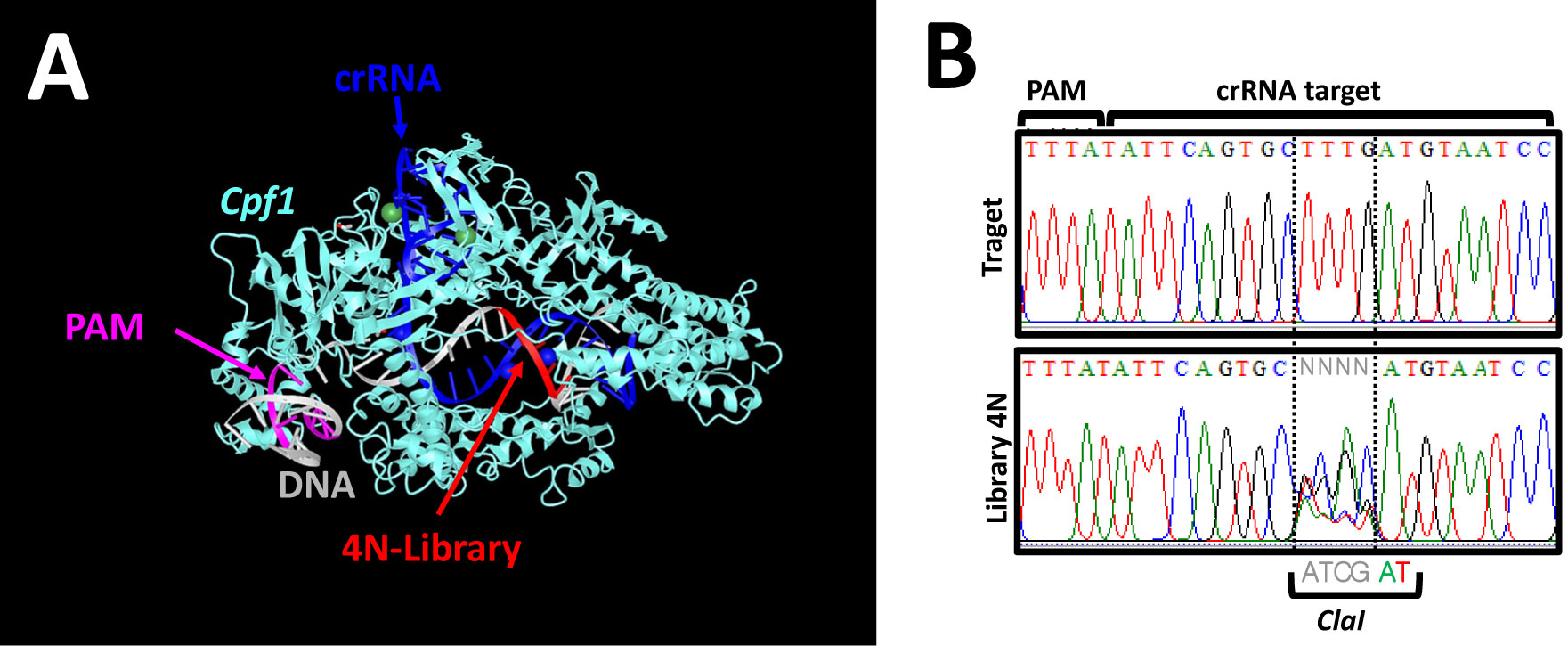
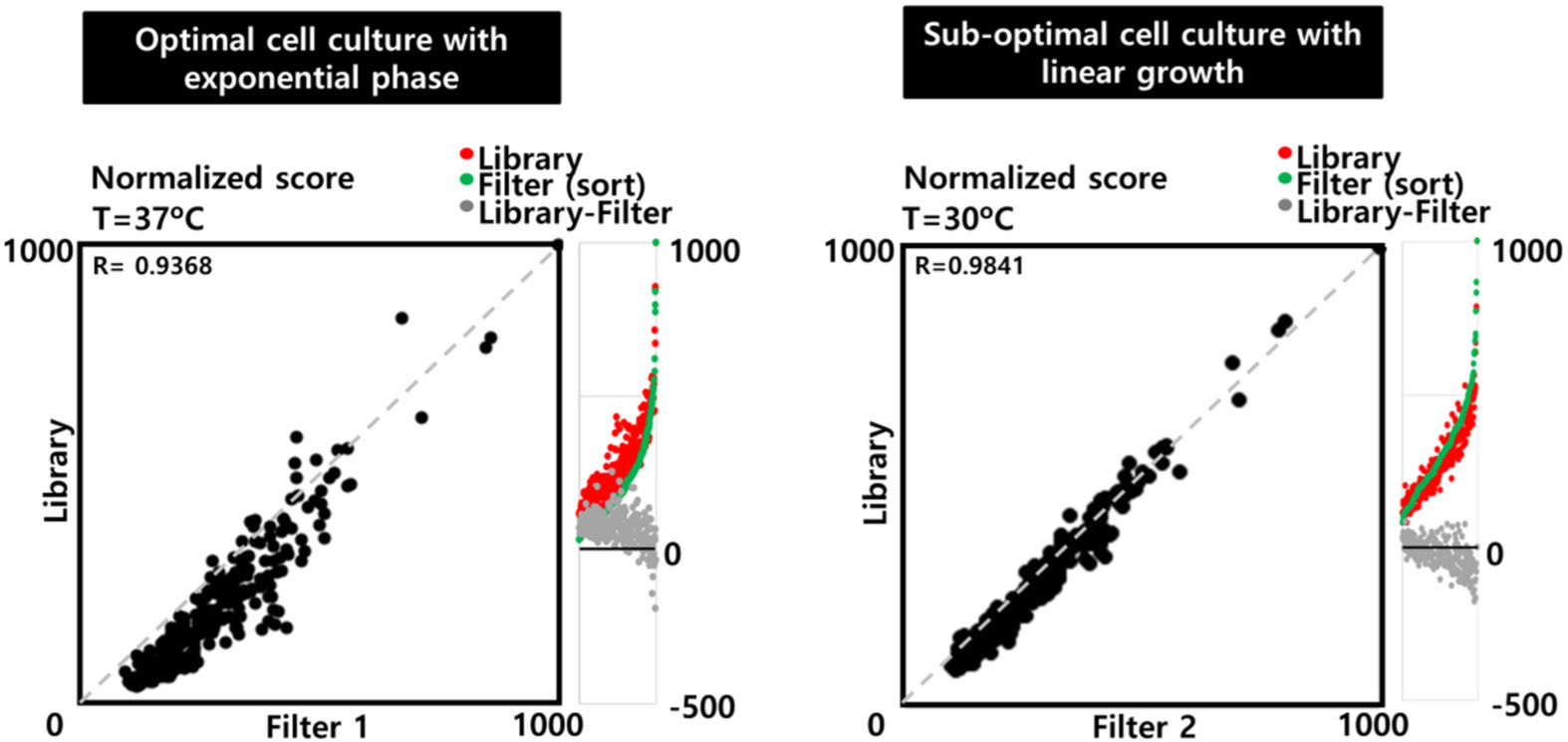
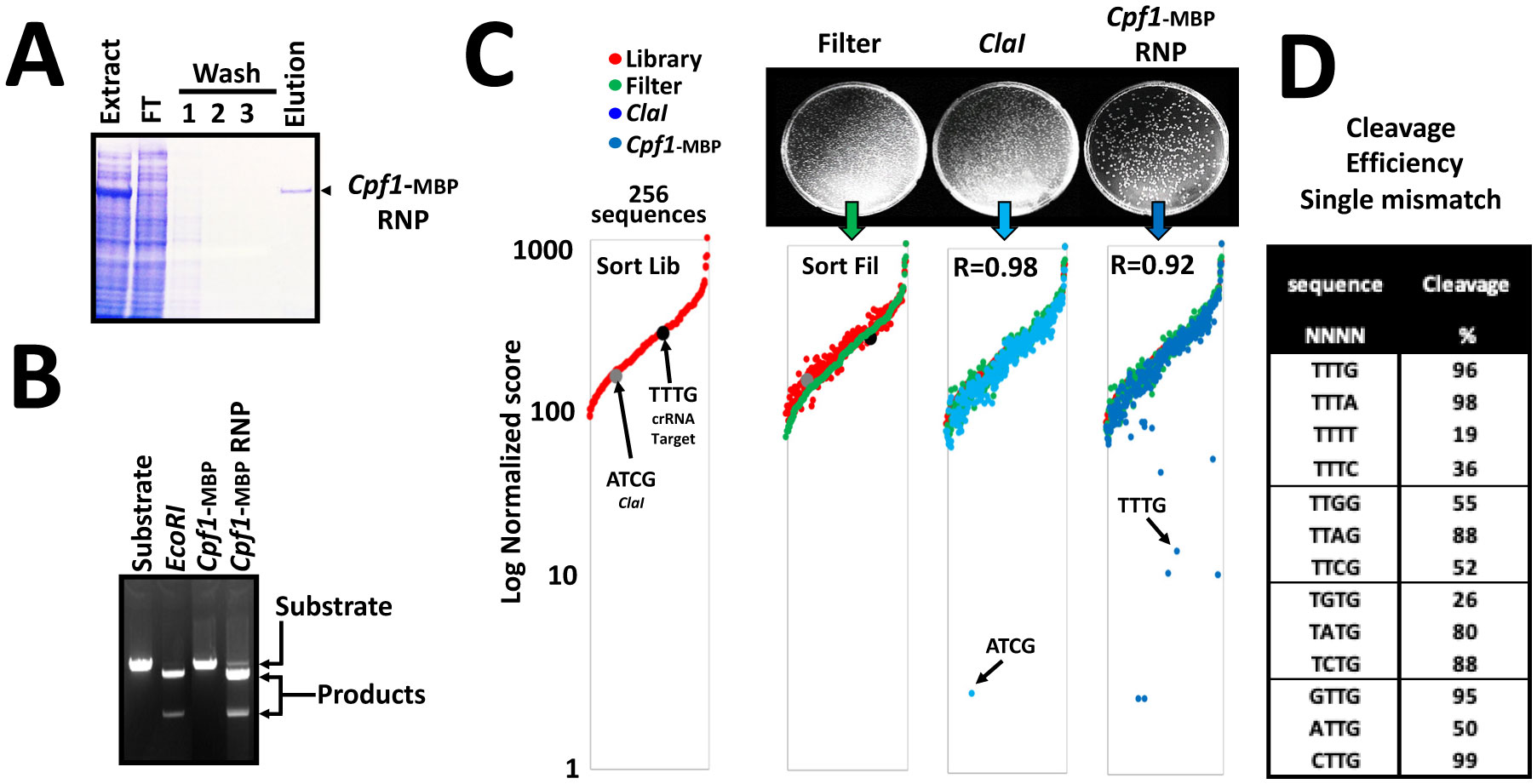
Nucleases currently used in genome engineering induce hydrolysis of DNA phosphate backbone in a sequence-specific manner. The RNA guided nucleases describe today are recognizing a sequence with two distinct molecular interactions: first, like a restriction endonuclease, by direct interaction between the protein and the DNA; and second, by hybridization of the guide RNA with the target DNA sequence. Here we report an in vitro assay to assess the cleavage specificity and the selectivity of the nucleases. The assay is designed using a plasmid encompassing the DNA target site degenerated at positions determined on structural feature. The results demonstrate that the Cpf1 RNA guided nuclease is highly specific for the target sequence, nevertheless its substrate selectivity is low compare to a restriction endonuclease.
Citation: Vincent Brondani. In vitro analysis of site specific nuclease selectivity by NGS[J]. AIMS Bioengineering, 2021, 8(4): 235-242. doi: 10.3934/bioeng.2021020
Nucleases currently used in genome engineering induce hydrolysis of DNA phosphate backbone in a sequence-specific manner. The RNA guided nucleases describe today are recognizing a sequence with two distinct molecular interactions: first, like a restriction endonuclease, by direct interaction between the protein and the DNA; and second, by hybridization of the guide RNA with the target DNA sequence. Here we report an in vitro assay to assess the cleavage specificity and the selectivity of the nucleases. The assay is designed using a plasmid encompassing the DNA target site degenerated at positions determined on structural feature. The results demonstrate that the Cpf1 RNA guided nuclease is highly specific for the target sequence, nevertheless its substrate selectivity is low compare to a restriction endonuclease.
CRISPR-associated protein
CRISPR from
Clustered regularly interspaced short palindromic repeat
CRISPR RNA
Double strand Break
Dulbecco's modified Eagle Medium
Dimethyl sulfoxide
Maltose binding protein
Next generation sequencing
Protospacer adjacent motif
transcription activator-like effector nucleases
Zinc finger nuclease

| [1] |
Kühnlein U, Linn S, Arber W (1969) Host specificity of DNA produced by Escherichia coli, XI. In vitro modification of phage fd replicative form. Proc Natl Acad Sci USA 63: 556-562. doi: 10.1073/pnas.63.2.556

|
| [2] |
Bibikova M, Carroll D, Segal DJ, et al. (2001) Stimulation of homologous recombination through targeted cleavage by chimeric nucleases. Mol Cell Biol 21: 289-297. doi: 10.1128/MCB.21.1.289-297.2001
|
| [3] |
Christian M, Cermak T, Doyle EL, et al. (2010) Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 186: 757-761. doi: 10.1534/genetics.110.120717
|
| [4] |
Gasiunas G, Barrangou R, Horvath P, et al. (2012) Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci USA 109: E2579-E2586. doi: 10.1073/pnas.1208507109
|
| [5] |
Jinek M, Chylinski K, Fonfara I, et al. (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337: 816-821. doi: 10.1126/science.1225829
|
| [6] |
Zetsche B, Gootenberg JS, Abudayyeh OO, et al. (2015) Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163: 759-771. doi: 10.1016/j.cell.2015.09.038
|
| [7] |
Nishimasu H, Ran FA, Hsu PD, et al. (2014) Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 156: 935-949. doi: 10.1016/j.cell.2014.02.001
|
| [8] |
Yamano T, Nishimasu H, Zetsche B, et al. (2016) Crystal structure of Cpf1 in complex with guide RNA and target DNA. Cell 165: 949-962. doi: 10.1016/j.cell.2016.04.003
|
| [9] |
Kim D, Kim J, Hur JK, et al. (2016) Genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells. Nat Biotechnol 34: 863-868. doi: 10.1038/nbt.3609
|
| [10] |
Kamps-Hughes N, Quimby A, Zhu Z, et al. (2013) Massively parallel characterization of restriction endonucleases. Nucleic Acids Res 41: e119. doi: 10.1093/nar/gkt257
|
| [11] |
Murugan K, Seetharam AS, Severin AJ, et al. (2020) CRISPR-Cas12a has widespread off-target and dsDNA-nicking effects: EDITORS'PICK: Cas12a nickase activities. J Biol Chem 295: 5538-5553. doi: 10.1074/jbc.RA120.012933
|
Figures(3)

Vincent Brondani. In vitro analysis of site specific nuclease selectivity by NGS[J]. AIMS Bioengineering, 2021, 8(4): 235-242. doi: 10.3934/bioeng.2021020






 DownLoad:
DownLoad: