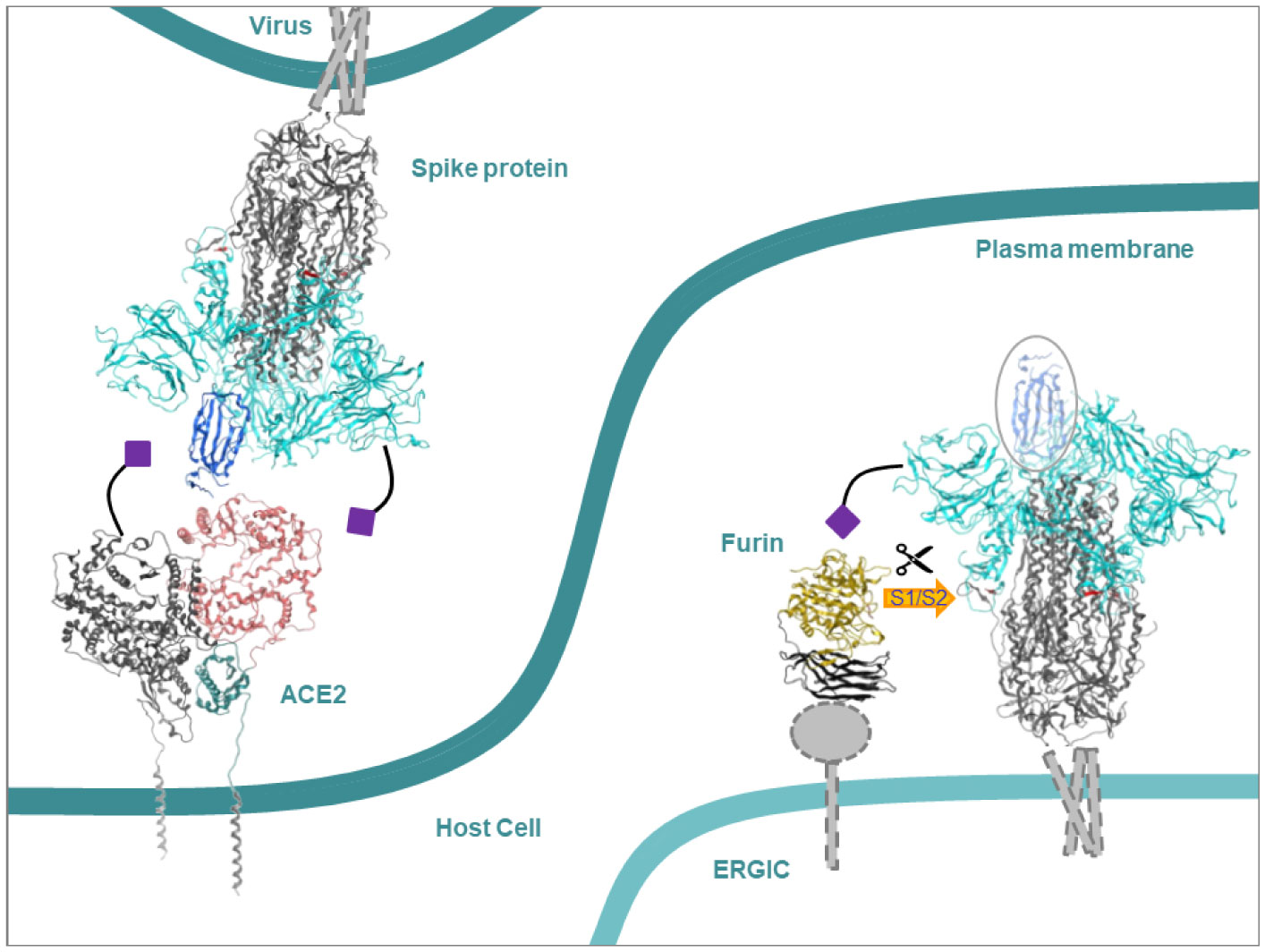
The initial step of interaction of some pathogens with the host is driven by the interaction of glycoproteins of either side via endcaps of their glycans. These end caps consist of sialic acids or sugar molecules. Coronaviruses (CoVs), including severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), are found to use this route of interaction. The strength and spatial interactions on the single molecule level of sialic acids with either the spike (S) protein of SARS coronaviruses, or human angiotensin-converting enzyme 2 (ACE2) and furin are probed and compared to the binding modes of those sugar molecules which are present in glycans of glycoproteins. The protocol of using single molecules is seen as a simplified but effective mimic of the complex mode of interaction of the glycans. Averaged estimated binding energies from a docking approach result in preferential binding of the sialic acids to a specific binding site of the S protein of human coronavirus OC43 (HCoV-OC43). Furin is proposed to provide better binding sites for sialic acids than ACE2, albeit outweighed by sites for other sugar molecules. Absolute minimal estimated binding energies indicate weak binding affinities and are indifferent to the type of sugar molecules and the proteins. Neither the proposed best binding sites of the sialic acids nor those of the sugar molecules overlap with any of the cleavage sites at the S protein and the active sites of the human proteins.
Citation: Chia-Wen Wang, Oscar K. Lee, Wolfgang B. Fischer. Screening coronavirus and human proteins for sialic acid binding sites using a docking approach[J]. AIMS Biophysics, 2021, 8(3): 248-263. doi: 10.3934/biophy.2021019
The initial step of interaction of some pathogens with the host is driven by the interaction of glycoproteins of either side via endcaps of their glycans. These end caps consist of sialic acids or sugar molecules. Coronaviruses (CoVs), including severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), are found to use this route of interaction. The strength and spatial interactions on the single molecule level of sialic acids with either the spike (S) protein of SARS coronaviruses, or human angiotensin-converting enzyme 2 (ACE2) and furin are probed and compared to the binding modes of those sugar molecules which are present in glycans of glycoproteins. The protocol of using single molecules is seen as a simplified but effective mimic of the complex mode of interaction of the glycans. Averaged estimated binding energies from a docking approach result in preferential binding of the sialic acids to a specific binding site of the S protein of human coronavirus OC43 (HCoV-OC43). Furin is proposed to provide better binding sites for sialic acids than ACE2, albeit outweighed by sites for other sugar molecules. Absolute minimal estimated binding energies indicate weak binding affinities and are indifferent to the type of sugar molecules and the proteins. Neither the proposed best binding sites of the sialic acids nor those of the sugar molecules overlap with any of the cleavage sites at the S protein and the active sites of the human proteins.

| [1] | Varki A (2008) Sialic acids in human health and disease. Trends Mol Med 14: 351-360. |
| [2] | Stencel-Baerenwald JE, Reiss K, Reiter DM, et al. (2014) The sweet spot: defining virus-sialic acid interactions. Nat Rev Microbiol 12: 739-749. |
| [3] | Koehler M, Delguste M, Sieben C, et al. (2020) Initial step of virus entry: virion binding to cell-surface glycans. Annu Rev Virol 7: 143-165. |
| [4] | Moscona A, Peluso RW (1993) Relative affinity of the human parainfluenza virus type 3 hemagglutinin-neuraminidase for sialic acid correlates with virus-induced fusion activity. J Virol 67: 6463-6468. |
| [5] | Viswanathan K, Chandrasekaran A, Srinivasan A, et al. (2010) Glycans as receptors for influenza pathogenesis. Glycoconj J 27: 561-570. |
| [6] | Sun XL (2021) The role of cell surface sialic acids for SARS-CoV-2 infection. Glycobiology online ahead of print. |
| [7] | Nicholis JM, Moss RB, Haslam SM (2013) The use of sialidase therapy for respiratory viral infections. Antivir Res 98: 401-409. |
| [8] | Balzarini J (2007) Targeting the glycans of glycoproteins: a novel paradigm for antiviral therapy. Nat Rev Microbiol 5: 583-597. |
| [9] | Colpitts CC, Schang LM (2014) A small molecule inhibits virion attachment to heparan sulfate- or sialic acid-containing glycans. J Virol 88: 7806-7817. |
| [10] | Rustmeier NH, Strebl M, Stehle T (2019) The symmetry of viral sialic acid binding sites—implications for antiviral strategies. Viruses 11: 947. |
| [11] | Heida R, Bhide YC, Gasbarri M, et al. (2020) Advances in the development of entry inhibitors for sialic-acid-targeting viruses. Drug Discov Today 26: 122-137. |
| [12] | Li F (2016) Structure, function, and evolution of coronavirus spike proteins. Annu Rev Virol 3: 237-261. |
| [13] | Bosch BJ, Van der Zee R, De Haan CAM, et al. (2003) The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J Virol 77: 8801-8811. |
| [14] | Watanabe Y, Allen JD, Wrapp D, et al. (2020) Site-specific glycan analysis of the SARS-CoV-2 spike. Science 369: 330-333. |
| [15] | Millet JK, Whittaker GR (2014) Host cell entry of middle east respiratory syndrome coronavirus after two-step, furin-mediated activation of the spike protein. P Natl Acad Sci USA 111: 15214-15219. |
| [16] | Coutard B, Valle C, de Lamballerie X, et al. (2020) The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antiviral Res 176: 104742. |
| [17] | Tang T, Bidon M, Jaimes JA, et al. (2020) Coronavirus membrane fusion mechanism offers a potential target for antiviral development. Antivir Res 178: 104792. |
| [18] | Li F, Li W, Farzan M, et al. (2005) Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 309: 1864-1868. |
| [19] | Mathewson AC, Bishop A, Yao Y, et al. (2008) Interaction of severe acute respiratory syndrome-coronavirus and NL63 coronavirus spike proteins with angiotensin converting enzyme-2. J Gen Virol 89: 2741-2745. |
| [20] | Tortorici MA, Walls AC, Lang Y, et al. (2019) Structural basis for human coronavirus attachment to sialic acid receptors. Nat Struct Mol Biol 26: 481-489. |
| [21] | Hulswit RJG, Lang Y, Bakkers MJG, et al. (2019) Human coronaviruses OC43 and HKU1 bind to 9-O-acetylated sialic acids via a conserved receptor-binding site in spike protein domain A. P Natl Acad Sci USA 116: 2681-2690. |
| [22] | Hao W, Ma B, Li Z, et al. (2021) Binding of the SARS-CoV-2 spike protein to glycans. Sci Bull 66: 1205-1214. |
| [23] | Baker AN, Richards SJ, Guy CS, et al. (2020) The SARS-COV-2 spike protein binds sialic acids and enables rapid detection in a lateral flow point of care diagnostic device. ACS Cent Sci 6: 2046-2052. |
| [24] | Awasthi M, Gulati S, Sarkar DP, et al. (2020) The sialoside-binding pocket of SARS-CoV-2 spike glycoprotein structurally resembles MERS-CoV. Viruses 12: 909. |
| [25] | Wu C, Zheng M, Yang Y, et al. (2020) Furin, a potential therapeutic target for COVID-19. iScience 23: 101642. |
| [26] | Thomas G (2002) Furin at the cutting edge: from protein traffic to embrygenesis and disease. Nat Rev Mol Cell Biol 3: 753-766. |
| [27] | Anderson ED, Molloy SS, Jean F, et al. (2002) The ordered and compartment-specific autoproteolytic removal of the furin intramolecular chaperone is required for enzyme activation. J Biol Chem 277: 12879-12890. |
| [28] | Shang J, Wan Y, Luo C, et al. (2020) Cell entry mechanisms of SARS-CoV-2. P Natl Acad Sci USA 117: 11727-11734. |
| [29] | Turner AJ, Hiscox JA, Hooper NM (2004) ACE2: From vasopeptidase to SARS virus receptor. Trends Pharmacol Sci 25: 291-294. |
| [30] | Li Y, Zhou W, Yang L, et al. (2020) Physiological and pathological regulation of ACE2, the SARS-CoV-2 receptor. Pharmacol Res 157: 104833. |
| [31] | Hoffmann M, Kleine-Weber H, Schroeder S, et al. (2020) SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181: 271-280. |
| [32] | Allen JD, Watanabe Y, Chawla H, et al. (2021) Subtle influence of ACE2 glycan processing on SARS-CoV-2 recognition. J Mol Biol 433: 166762. |
| [33] | Mysinger MM, Carchia M, Irwin JJ, et al. (2012) Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. J Med Chem 55: 6582-6594. |
| [34] | Bursulaya BD, Totrov M, Abagyan R, et al. (2003) Comparative study of several algorithms for flexible ligand docking. J Comput Aid Mol Des 17: 755-763. |
| [35] | Rozano L, Abdullah Zawawi MR, Ahmad MA, et al. (2017) Computational analysis of Gynura bicolor bioactive compounds as dipeptidyl peptidase-IV inhibitor. Adv Bioinform 5124165. |
| [36] | Kellenberger E, Rodrigo J, Muller P, et al. (2004) Comparative evaluation of eight docking tools for docking and virtual screening accuracy. Proteins 57: 225-242. |
| [37] | Chen H, Lyne PD, Giordanetto F, et al. (2006) On evaluating molecular-docking methods for pose prediction and enrichment factors. J Chem Inf Model 46: 401-415. |
| [38] | Nurisso A, Kozmon S, Imberty A (2008) Comparison of docking methods for carbohydrate binding in calcium-dependent lectins and prediction of the carbohydrate binding mode to sea cucumber lectin CEL-III. Mol Simulat 34: 469-479. |
| [39] | Reulecke I, Lange G, Albrecht J, et al. (2008) Towards an integrated description of hydrogen bonding and dehydration: decreasing false positives in virtual screening with the HYDE scoring function. Chem Med Chem 3: 885-897. |
| [40] | Bakker TR, Piperi C, Davies EA, et al. (2002) Comparison of CD22 binding to native CD45 and synthetic oligosaccharide. Eur J Immunol 32: 1924-1932. |
| [41] | Crocker PR, Blixt O, Collins BE, et al. (2003) Sialoside specificity of the siglec family assessed using novel multivalent probes: identification of potent inhibitors of myelin-associated glycoprotein. J BiolChem 278: 31007-31019. |
| [42] | Yamakawa N, Yasuda Y, Yoshimura A, et al. (2020) Discovery of a new sialic acid binding region that regulates Siglec-7. Sci Rep 10: 1-14. |
| [43] | Li W, Hulswit RJG, Widjaja I, et al. (2017) Identification of sialic acid-binding function for the Middle East respiratory syndrome coronavirus spike glycoprotein. Proc Natl Acad Sci USA 114: E8508-E8517. |
| [44] | Shajahan A, Archer-Hartmann S, Supekar NT, et al. (2020) Comprehensive characterization of N-and O-glycosylation of SARS-CoV-2 human receptor angiotensin converting enzyme 2. Glycobiology 31: 410-424. |
| [45] | Mamedov T, Musayeva I, Acsora R, et al. (2019) Engineering, and production of functionally active human Furin in N. benthamiana plant: In vivo post-translational processing of target proteins by Furin in plants. PLoS One 14: e0213438. |
| [46] | Chen Y, Guo Y, Pan Y, et al. (2020) Structure analysis of the receptor binding of 2019-nCoV. Biophys Res Comm 525: 135-140. |
| [47] | Krokhin O, Li Y, Andonov A, et al. (2003) Mass spectrometric characterization of proteins from the SARS virus: a preliminary report. Mol Cell Proteomics 2: 346-356. |
| [48] | Fung T S, Liu D X (2018) Post-translational modifications of coronavirus proteins: roles and function. Future Virol 13: 405-430. |
 biophy-08-03-019-s001.pdf biophy-08-03-019-s001.pdf |
 |




Figures(5) / Tables(1)

Chia-Wen Wang, Oscar K. Lee, Wolfgang B. Fischer. Screening coronavirus and human proteins for sialic acid binding sites using a docking approach[J]. AIMS Biophysics, 2021, 8(3): 248-263. doi: 10.3934/biophy.2021019







 DownLoad:
DownLoad: