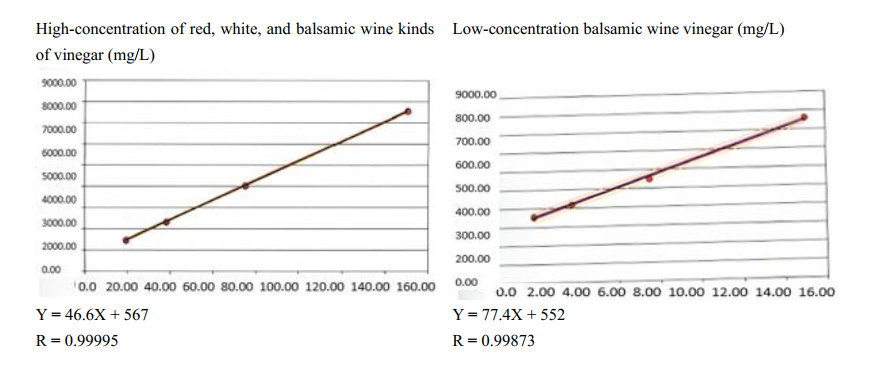
Sulfur dioxide is generally used in wine and vinegar production. It is employed to decrease the bacteria' growth, improve the wines' aroma (since it supports the extraction of polyphenols during maceration), protect the wines from chemical oxidation and the musts from chemical and enzymatic oxidation (blocking free radicals and oxidase enzymes such as tyrosinase and laccase). The composition and storage conditions (i.e., pH, temperature, and alcohol levels) affect oenological results. In various countries, competent authorities have imposed legal limits since it can have toxic effects on humans. It is crucial to dose SO2 levels to allow vinegar production and compliance with legal limits. The iodometric titration named "Ripper test" is the legal method used to dose it in vinegar. In this work, an automatized colorimetric test was validated using the international guidelines ISO/IEC (2017) to allow its use instead of the Ripper test. The test reliability was verified on white, red, and balsamic vinegar with low or high SO2 content. The automatized test showed linearity, precision, and reproducibility similar to the Ripper test, but the accuracy parameter was not respected for the vinegar with a low concentration of SO2. Therefore, the automatized colorimetric test can be helpful to dose SO2 in vinegar with high concentrations of SO2.
Citation: Irene Dini, Antonello Senatore, Daniele Coppola, Andrea Mancusi. Validation of a rapid test to dose SO2 in vinegar[J]. AIMS Agriculture and Food, 2023, 8(1): 1-24. doi: 10.3934/agrfood.2023001
Sulfur dioxide is generally used in wine and vinegar production. It is employed to decrease the bacteria' growth, improve the wines' aroma (since it supports the extraction of polyphenols during maceration), protect the wines from chemical oxidation and the musts from chemical and enzymatic oxidation (blocking free radicals and oxidase enzymes such as tyrosinase and laccase). The composition and storage conditions (i.e., pH, temperature, and alcohol levels) affect oenological results. In various countries, competent authorities have imposed legal limits since it can have toxic effects on humans. It is crucial to dose SO2 levels to allow vinegar production and compliance with legal limits. The iodometric titration named "Ripper test" is the legal method used to dose it in vinegar. In this work, an automatized colorimetric test was validated using the international guidelines ISO/IEC (2017) to allow its use instead of the Ripper test. The test reliability was verified on white, red, and balsamic vinegar with low or high SO2 content. The automatized test showed linearity, precision, and reproducibility similar to the Ripper test, but the accuracy parameter was not respected for the vinegar with a low concentration of SO2. Therefore, the automatized colorimetric test can be helpful to dose SO2 in vinegar with high concentrations of SO2.

| [1] |
Abaje IB, Bello Y, Ahmad SA (2020) A review of air quality and concentrations of air pollutants in Nigeria. JASEM 24: 373–379. https://doi.org/10.4314/jasem.v24i2.25 doi: 10.4314/jasem.v24i2.25

|
| [2] | Schroeter LC (1966) Sulfur dioxide: Applications in foods, beverages, and pharmaceuticals, Long Island City: Pergamon Press, Inc. |
| [3] |
Mandrile L, Cagnasso I, Berta L, et al. (2020) Direct quantification of sulfur dioxide in wine by Surface Enhanced Raman Spectroscopy. Food Chem 326: 127009. https://doi.org/10.1016/j.foodchem.2020.127009 doi: 10.1016/j.foodchem.2020.127009
|
| [4] |
Morgan SC, Tantikachornkiat M, Scholl CM, et al. (2019) The effect of sulfur dioxide addition at crush on the fungal and bacterial communities and the sensory attributes of Pinot gris wines. Int J Food Microbiol 290: 1–14. https://doi.org/10.1016/j.ijfoodmicro.2018.09.020 doi: 10.1016/j.ijfoodmicro.2018.09.020
|
| [5] |
Gabriele M, Gerardi C, Lucejko JJ, et al. (2018) Effects of low sulfur dioxide concentrations on bioactive compounds and antioxidant properties of Aglianico red wine. Food Chem 245: 1105–1112. https://doi.org/10.1016/j.foodchem.2017.11.060 doi: 10.1016/j.foodchem.2017.11.060
|
| [6] | Giacosa S, Segade SR, Cagnasso E, et al. (2019) Red Wine Technology, 1 Eds., New York: Academic Press, 309–321. https://doi.org/10.1016/B978-0-12-814399-5.00021-9 |
| [7] |
Seralini G, Douzelet J, Halley J (2021) Sulfur in Wines and Vineyards: Taste and Comparative Toxicity to Pesticides. Food Nutr J 6: 231. https://doi.org/10.29011/2575-7091.100131 doi: 10.29011/2575-7091.100131
|
| [8] | Directive 2003/89/EC of the European Parliament and of the Council of 10 November 2003 amending Directive 2000/13/EC as regards indication of the ingredients present in foodstuffs (Text with EEA relevance). (2003) Available from: https://eur-lex.europa.eu/legal-content/en/ALL/?uri=CELEX%3A32003L008913/12/2014. |
| [9] | Commission Regulation No 606/2009 of 10 July 2009. Laying down certain detailed rules for implementing Council Regulation (EC) No 479/2008 as regards the categories of grapevine products, oenological practices and the applicable restrictions. (2009) Available from: https://eur-lex.europa.eu/eli/reg/2009/606/oj/eng. |
| [10] |
Jenkins TW, Howe PA, Sacks GL, et al. (2020) Determination of molecular and " Truly" free sulfur dioxide in wine: a comparison of headspace and conventional methods. AJEV 71: 222–230. https://doi.org/10.5344/ajev.2020.19052 doi: 10.5344/ajev.2020.19052
|
| [11] |
Danchana K, Clavijo S, Cerdà V (2019) Conductometric determination of sulfur dioxide in wine using a multipumping system coupled to a gas-diffusion cell. Anal Lett 52: 1363–1378. https://doi.org/10.1080/00032719.2018.1539742 doi: 10.1080/00032719.2018.1539742
|
| [12] | Eurachem Guide (2014) Eurachem Guide: The Fitness for Purpose of Analytical Methods—A Laboratory Guide to Method Validation and Related Topics, 2 Eds., Available from: https://www.eurachem.org/images/stories/Guides/pdf/MV_guide_2nd_ed_EN.pdf. |
| [13] |
Pepe P, Bosco A, Capuano F, et al. (2021) Towards an Integrated Approach for Monitoring Toxoplasmosis in Southern Italy. Animals 1: 1949. https://doi.org/10.3390/ani11071949 doi: 10.3390/ani11071949
|
| [14] |
Laneri S, Di Lorenzo R, Sacchi A, et al. (2019) Dosage of bioactive molecules in the nutricosmeceutical helix aspersa muller mucus and formulation of new cosmetic cream with moisturizing effect. Nat Prod Commun 14: 1–7. https://doi.org/10.1177/1934578X19868606 doi: 10.1177/1934578X19868606
|
| [15] |
Mancusi F, Capuano S, Girardi O, et al. (2022) Detection of SARS-CoV-2 RNA in bivalve mollusks by droplet digital RT-PCR (dd RT-PCR). Int J Environ Res Public Health 19: 943. https://doi.org/10.3390/ijerph19020943 doi: 10.3390/ijerph19020943
|
| [16] |
Fanelli F, Cozzi G, Raiola A, et al. (2017) Raisins and currants as conventional nutraceuticals in Italian market: Natural occurrence of Ochratoxin A. J Food Sci 82: 2306–2312. https://doi: 10.1111/1750-3841.13854 doi: 10.1111/1750-3841.13854
|
| [17] |
Mancusi A, Giordano A, Bosco A, et al. (2022) Development of a droplet digital polymerase chain reaction tool for the detection of Toxoplasma gondii in meat samples. Parasitol Res 121: 1467–1473. https://doi.org/10.1007/s00436-022-07477-9 doi: 10.1007/s00436-022-07477-9
|
| [18] |
Capuano F, Capparelli R, Mancusi A, et al. (2013) Detection of Brucella spp. in Stretched Curd Cheese as Assessed by Molecular Assays. J Food Saf 33: 145–148. https://doi.org/10.1111/jfs.12034 doi: 10.1111/jfs.12034
|
| [19] |
Cristiano D, Peruzy MF, Aponte M, et al. (2021) Comparison of droplet digital PCR vs real-time PCR for Yersinia enterocolitica detection in vegetables. Int J Food Microbiol 354: 109321. https://doi.org/10.1016/j.ijfoodmicro.2021.109321 doi: 10.1016/j.ijfoodmicro.2021.109321
|
| [20] | OIV 2007 Organisation Internationale de la Vigne et du Vin (2007) Available from: https://www.oiv.int/en/technical-standards-and-documents/methods-of-analysis/compendium-of-international-methods-of-analysis-of-wines-and-musts. |
| [21] |
Shapiro SS, Wilk MB (1965) An analysis of variance test for normality (complete samples). Biometrika 52: 591–611. https://doi.org/10.2307/2333709 doi: 10.2307/2333709
|
| [22] | Huber PJ (1981) Robust statistics, New York: Wiley, 1–384. |
| [23] |
Dini I, Seccia S, Senatore A, et al. (2019) Development and validation of an analytical method for total polyphenols quantification in extra virgin olive oils. Food Anal Met 13: 457–464. https://doi.org/10.1007/s12161-019-01657-7 doi: 10.1007/s12161-019-01657-7
|
| [24] |
Dini I, Di Lorenzo R, Senatore A, et al. (2020) Validation of rapid enzymatic quantification of acetic acid in vinegar on automated spectrophotometric system. Foods 9: 761. https://doi.org/10.3390/foods9060761 doi: 10.3390/foods9060761
|
| [25] | ISO/IEC 17025: 2017 General requirements for the competence of testing and calibration laboratories (2017) Available from: https://www.iso.org/standard/66912.html. |
| [26] |
Molognoni L, Daguer H, Dos Santos IR, et al. (2019) Influence of method validation parameters in the measurement uncertainty estimation by experimental approaches in food preservatives analysis. Food Chem 282: 147–152. https://doi.org/10.1016/j.foodchem.2018.12.115 doi: 10.1016/j.foodchem.2018.12.115
|
| [27] |
Dini I, Di Lorenzo R, Senatore A, et al. (2021) Comparison between Mid-Infrared (ATR-FTIR) spectroscopy and official analysis methods for determination of the concentrations of alcohol, SO2, and total acids in wine. Separations 8: 191. https://doi.org/10.3390/separations8100191 doi: 10.3390/separations8100191
|

Figures(2) / Tables(18)

Irene Dini, Antonello Senatore, Daniele Coppola, Andrea Mancusi. Validation of a rapid test to dose SO2 in vinegar[J]. AIMS Agriculture and Food, 2023, 8(1): 1-24. doi: 10.3934/agrfood.2023001







 DownLoad:
DownLoad: