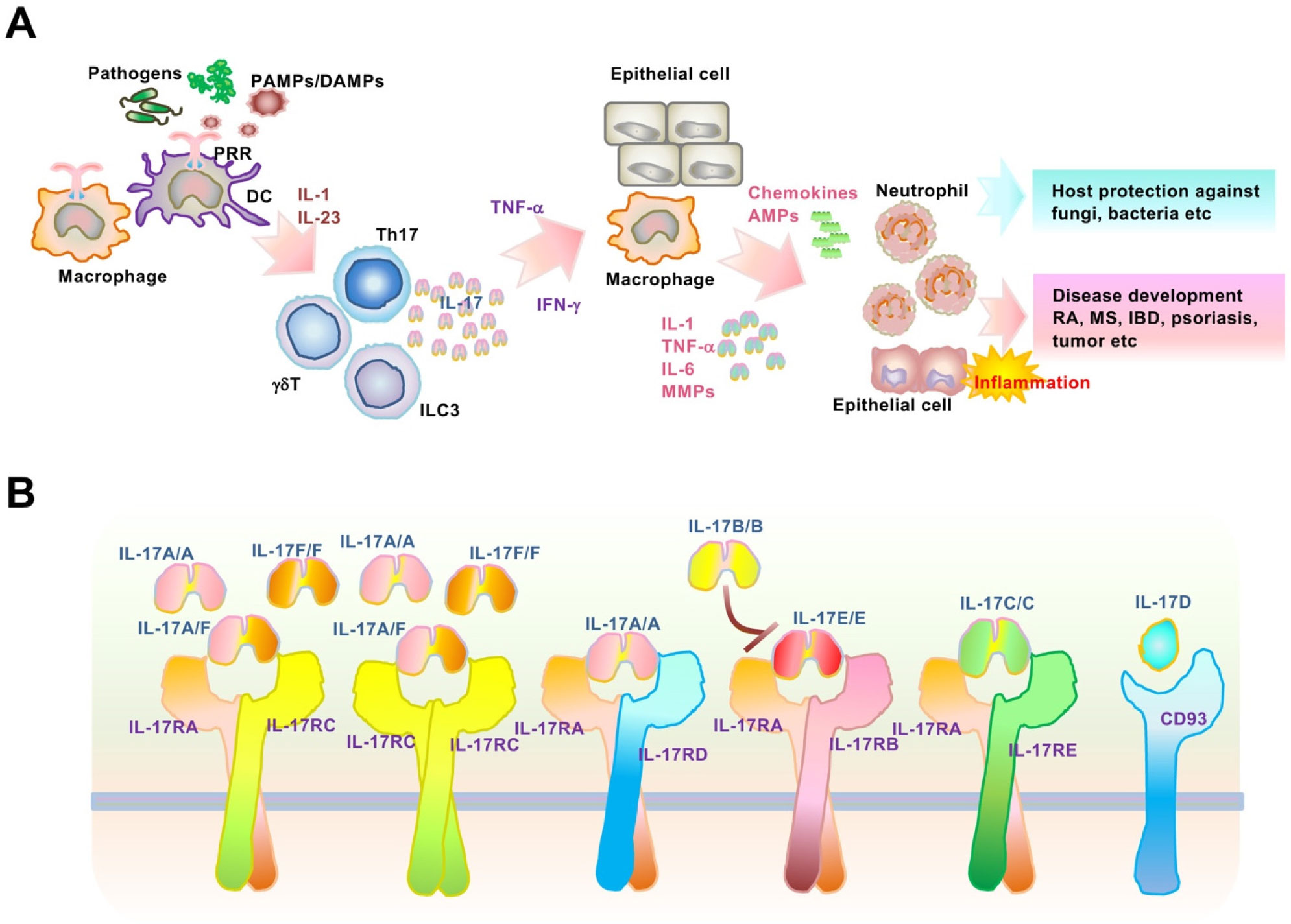
Interleukin (IL)-17 is a proinflammatory cytokine mainly produced by immune cells, especially activated T-helper 17 cells, which contribute to chronic inflammatory and autoimmune diseases including psoriasis. Although the molecular mechanisms of transcription in IL-17-mediated signaling pathways are well established, post-transcriptional control remains to be elucidated. Notably, IL-17 regulates post-transcriptional modifications, which induce elevated levels of target inflammatory mRNAs. Regnase-1, an endoribonuclease and deubiquitinase, post-transcriptionally downregulates various IL-17-driven signaling pathways, including mRNA stability. The ACT1-TBK1/IKKϵ pathway and ARID5A were induced and activated by IL-17-stimulation, leading to the inhibition of inflammatory mRNA degradation by Regnase-1. In this review, we focus on IL-17-mediated mRNA stabilization of psoriasis-related IκB-ζ and provide novel therapeutic strategies for the treatment of Th17-mediated inflammation and autoimmunity.
Citation: Ryuta Muromoto, Kenji Oritani, Tadashi Matsuda. IL-17 signaling is regulated through intrinsic stability control of mRNA during inflammation[J]. AIMS Allergy and Immunology, 2022, 6(3): 188-199. doi: 10.3934/Allergy.2022014
Interleukin (IL)-17 is a proinflammatory cytokine mainly produced by immune cells, especially activated T-helper 17 cells, which contribute to chronic inflammatory and autoimmune diseases including psoriasis. Although the molecular mechanisms of transcription in IL-17-mediated signaling pathways are well established, post-transcriptional control remains to be elucidated. Notably, IL-17 regulates post-transcriptional modifications, which induce elevated levels of target inflammatory mRNAs. Regnase-1, an endoribonuclease and deubiquitinase, post-transcriptionally downregulates various IL-17-driven signaling pathways, including mRNA stability. The ACT1-TBK1/IKKϵ pathway and ARID5A were induced and activated by IL-17-stimulation, leading to the inhibition of inflammatory mRNA degradation by Regnase-1. In this review, we focus on IL-17-mediated mRNA stabilization of psoriasis-related IκB-ζ and provide novel therapeutic strategies for the treatment of Th17-mediated inflammation and autoimmunity.

| [1] |
Mosmann TR, Coffman RL (1989) TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol 7: 145-173. https://doi.org/10.1146/annurev.iy.07.040189.001045 
|
| [2] |
Korn T, Bettelli E, Oukka M, et al. (2009) IL-17 and Th17 Cells. Annu Rev Immunol 27: 485-517. https://doi.org/10.1146/annurev.immunol.021908.132710
|
| [3] |
Chung SH, Ye XQ, Iwakura Y, et al. (2021) Interleukin-17 family members in health and disease. Int Immunol 33: 723-729. https://doi.org/10.1093/intimm/dxab075
|
| [4] | Mills KHG (2022) IL-17 and IL-17-producing cells in protection versus pathology. Nat Rev Immunol 2022: 1-17. https://doi.org/10.1038/s41577-022-00746-9 |
| [5] |
Lee GR (2018) The balance of Th17 versus Treg cells in autoimmunity. Int J Mol Sci 19: 730. https://doi.org/10.3390/ijms19030730
|
| [6] |
Milovanovic J, Arsenijevic A, Stojanovic B, et al. (2020) Interleukin-17 in chronic inflammatory neurological diseases. Front Immunol 11: 947. https://doi.org/10.3389/fimmu.2020.00947
|
| [7] |
von Stebut E, Boehncke WH, Ghoreschi K, et al. (2020) IL-17A in psoriasis and beyond: Cardiovascular and metabolic implications. Front Immunol 10: 3096. https://doi.org/10.3389/fimmu.2019.03096
|
| [8] |
Akira S, Maeda K (2021) Control of RNA stability in immunity. Annu Rev Immunol 39: 481-509. https://doi.org/10.1146/annurev-immunol-101819-075147
|
| [9] |
Yoshinaga M, Takeuchi O (2019) Post-transcriptional control of immune responses and its potential application. Clin Transl Immunol 8: e1063. https://doi.org/10.1002/cti2.1063
|
| [10] |
Qian Y, Liu C, Hartupee J, et al. (2007) The adaptor Act1 is required for interleukin 17-dependent signaling associated with autoimmune and inflammatory disease. Nat Immunol 8: 247-256. https://doi.org/10.1038/ni1439
|
| [11] |
Onishi RM, Gaffen SL (2010) Interleukin-17 and its target genes: mechanisms of interleukin-17 function in disease. Immunology 129: 311-321. https://doi.org/10.1111/j.1365-2567.2009.03240.x
|
| [12] |
Tohyama M, Hanakawa Y, Shirakata Y, et al. (2009) IL-17 and IL-22 mediate IL-20 subfamily cytokine production in cultured keratinocytes via increased IL-22 receptor expression. Eur J Immunol 39: 2779-2788. https://doi.org/10.1002/eji.200939473
|
| [13] |
Anderson P (2009) Intrinsic mRNA stability helps compose the inflammatory symphony. Nat Immunol 10: 233-234. https://doi.org/10.1038/ni0309-233
|
| [14] |
Anderson P (2010) Post-transcriptional regulons coordinate the initiation and resolution of inflammation. Nat Rev Immunol 10: 24-35. https://doi.org/10.1038/nri2685
|
| [15] |
Masuda K, Ripley B, Nishimura R, et al. (2013) Arid5a controls IL-6 mRNA stability, which contributes to elevation of IL-6 level in vivo. P Natl Acad Sci USA 110: 9409-9414. https://doi.org/10.1073/pnas.1307419110
|
| [16] |
Matsushita K, Takeuchi O, Standley DM, et al. (2009) Zc3h12a is an RNase essential for controlling immune responses by regulating mRNA decay. Nature 458: 1185-1190. https://doi.org/10.1038/nature07924
|
| [17] |
Uehata T, Iwasaki H, Vandenbon A, et al. (2013) Malt1-induced cleavage of regnase-1 in CD4(+) helper T cells regulates immune activation. Cell 153: 1036-1049. https://doi.org/10.1016/j.cell.2013.04.034
|
| [18] |
Garg AV, Amatya N, Chen K, et al. (2015) MCPIP1 Endoribonuclease activity negatively regulates interleukin-17-mediated signaling and inflammation. Immunity 43: 475-487. https://doi.org/10.1016/j.immuni.2015.07.021
|
| [19] |
Amatya N, Childs EE, Cruz JA, et al. (2018) IL-17 integrates multiple self-reinforcing, feed-forward mechanisms through the RNA binding protein Arid5a. Sci Signal 11: eaat4617. https://doi.org/10.1126/scisignal.aat4617
|
| [20] |
Tanaka H, Arima Y, Kamimura D, et al. (2019) Phosphorylation-dependent Regnase-1 release from endoplasmic reticulum is critical in IL-17 response. J Exp Med 216: 1431-1449. https://doi.org/10.1084/jem.20181078
|
| [21] |
Bulek K, Liu C, Swaidani S, et al. (2011) The inducible kinase IKKi is required for IL-17-dependent signaling associated with neutrophilia and pulmonary inflammation. Nat Immunol 12: .844-852. https://doi.org/10.1038/ni.2080
|
| [22] |
Qu F, Gao H, Zhu S, et al. (2012) TRAF6-dependent Act1 phosphorylation by the IκB kinase-related kinases suppresses interleukin-17-induced NF-κB activation. Mol Cell Biol 32: 3925-3937. https://doi.org/10.1128/MCB.00268-12
|
| [23] |
Muromoto R, Hirao T, Tawa K, et al. (2016) IL-17A plays a central role in the expression of psoriasis signature genes through the induction of IκB-ζ in keratinocytes. Int Immunol 28: 443-452. https://doi.org/10.1093/intimm/dxw011
|
| [24] |
Willems M, Dubois N, Musumeci L (2016) IκBζ: an emerging player in cancer. Oncotarget 7: 66310-66322. https://doi.org/10.18632/oncotarget.11624
|
| [25] |
Bertelsen T, Ljungberg C, Kjellerup RB, et al. (2017) IL-17F regulates psoriasis-associated genes through IκBζ. Exp Dermatol 26: 234-241. https://doi.org/10.1111/exd.13182
|
| [26] |
Müller A, Hennig A, Lorscheid S, et al. (2018) IκBζ is a key transcriptional regulator of IL-36-driven psoriasis-related gene expression in keratinocytes. P Natl Acad Sci USA 115: 10088-10093. https://doi.org/10.1073/pnas.1801377115
|
| [27] |
Yamazaki S, Muta T, Matsuo S, et al. (2005) Stimulus-specific induction of a novel nuclear factor-κB regulator, IκB-ζ, via toll/interleukin-1 receptor is mediated by mRNA stabilization. J Biol Chem 280: 1678-1687. https://doi.org/10.1074/jbc.M409983200
|
| [28] |
Okuma A, Hoshino K, Ohba T, et al. (2013) Enhanced apoptosis by disruption of the STAT3-IκB-ζ signaling pathway in epithelial cells induces Sjögren's syndrome-like autoimmune disease. Immunity 38: 450-460. https://doi.org/10.1016/j.immuni.2012.11.016
|
| [29] |
Muromoto R, Tawa K, Ohgakiuchi Y, et al. IκB-ζ expression requires both TYK2/STAT3 activity and IL-17-regulated mRNA stabilization. Immunohorizons 3: 172-185. https://doi.org/10.4049/immunohorizons.1900023
|
| [30] |
Ohgakiuchi Y, Saino Y, Muromoto R, et al. (2020) Dimethyl fumarate dampens IL-17-ACT1-TBK1 axis-mediated phosphorylation of Regnase-1 and suppresses IL-17-induced IκB-ζ expression. Biochem Biophys Res Commun 521: 957-963. https://doi.org/10.1016/j.bbrc.2019.11.036
|
| [31] |
Nestle FO, Kaplan DH, Barker J (2009) Psoriasis. N Engl J Med 361: 496-509. https://doi.org/10.1056/NEJMra0804595
|
| [32] |
Christophers E, Metzler G, Röcken M (2014) Bimodal immune activation in psoriasis. Brit J Dermatol 170: 59-65. https://doi.org/10.1111/bjd.12631
|
| [33] |
Lowes MA, Suárez-Fariñas M, Krueger JG (2014) Immunology of psoriasis. Annu Rev Immunol 32: 227-255. https://doi.org/10.1146/annurev-immunol-032713-120225
|
| [34] |
Lynde CW, Poulin Y, Vender R, et al. (2014) Interleukin 17A: toward a new understanding of psoriasis pathogenesis. J Am Acad Dermatol 71: 141-150. https://doi.org/10.1016/j.jaad.2013.12.036
|
| [35] |
Baliwag J, Barnes DH, Johnston A (2015) Cytokines in psoriasis. Cytokine 73: 342-350. https://doi.org/10.1016/j.cyto.2014.12.014
|
| [36] |
Lorscheid S, Müller A, Löffler J, et al. (2019) Keratinocyte-derived IκBζ drives psoriasis and associated systemic inflammation. JCI Insight 4: e130835. https://doi.org/10.1172/jci.insight.130835
|
| [37] |
Mandal A, Kumbhojkar N, Reilly C, et al. (2020) Treatment of psoriasis with NFKBIZ siRNA using topical ionic liquid formulations. Sci Adv 6: eabb6049. https://doi.org/10.1126/sciadv.abb6049
|
| [38] |
Schwartz DM, Kanno Y, Villarino A, et al. (2017) JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat Rev Drug Discov 16: 843-862. https://doi.org/10.1038/nrd.2017.201
|
| [39] |
Muromoto R, Oritani K, Matsuda T (2022) Current understanding of the role of tyrosine kinase 2 signaling in immune responses. World J Biol Chem 13: 1-14. https://doi.org/10.4331/wjbc.v13.i1.1
|
| [40] |
Muromoto R, Shimoda K, Oritani K, et al. (2021) Therapeutic advantage of Tyk2 inhibition for Treating autoimmune and chronic inflammatory diseases. Biol Pharm Bull 44: 1585-1592. https://doi.org/10.1248/bpb.b21-00609
|
| [41] |
Burke JR, Cheng L, Gillooly KM, et al. (2019) Autoimmune pathways in mice and humans are blocked by pharmacological stabilization of the TYK2 pseudokinase domain. Sci Transl Med 11: eaaw1736. https://doi.org/10.1126/scitranslmed.aaw1736
|
| [42] |
Chang Y, Xu S, Ding K (2019) Tyrosine kinase 2 (TYK2) allosteric inhibitors to treat autoimmune diseases. J Med Chem 62: 8951-8952. https://doi.org/10.1021/acs.jmedchem.9b01612
|
| [43] |
Sadeghi A, Tahmasebi S, Mahmood A, et al. (2021) Th17 and Treg cells function in SARS-CoV2 patients compared with healthy controls. J Cell Physiol 236: 2829-2839. https://doi.org/10.1002/jcp.30047
|
| [44] |
Leija-Martínez JJ, Huang F, Del-Río-Navarro BE, et al. (2020) IL-17A and TNF-α as potential biomarkers for acute respiratory distress syndrome and mortality in patients with obesity and COVID-19. Med Hypotheses 144: 109935. https://doi.org/10.1016/j.mehy.2020.109935
|
| [45] |
Hasan MZ, Islam S, Matsumoto K, et al. (2021) SARS-CoV-2 infection initiates interleukin-17-enriched transcriptional response in different cells from multiple organs. Sci Rep 11: 16814. https://doi.org/10.1038/s41598-021-96110-3
|
| [46] |
Maslennikov R, Ivashkin V, Vasilieva E, et al. (2021) Interleukin 17 antagonist netakimab is effective and safe in the new coronavirus infection (COVID-19). Eur Cytokine Netw 32: 8-14. https://doi.org/10.1684/ecn.2021.0463
|
| [47] |
Avdeev SN, Trushenko NV, Tsareva NA, et al. (2021) Anti-IL-17 monoclonal antibodies in hospitalized patients with severe COVID-19: a pilot study. Cytokine 146: 155627. https://doi.org/10.1016/j.cyto.2021.155627
|

Figures(2)

Ryuta Muromoto, Kenji Oritani, Tadashi Matsuda. IL-17 signaling is regulated through intrinsic stability control of mRNA during inflammation[J]. AIMS Allergy and Immunology, 2022, 6(3): 188-199. doi: 10.3934/Allergy.2022014







 DownLoad:
DownLoad: