Citation: Junji Ito, Emanuele Lucrezia, Günther Palm, Sonja Grün. Detection and evaluation of bursts in terms of novelty and surprise[J]. Mathematical Biosciences and Engineering, 2019, 16(6): 6990-7008. doi: 10.3934/mbe.2019351

| [1] | K. D. Harris, H. Hirase, X. Leinekugel, et al., Temporal interaction between single spikes and complex spike bursts in hippocampal pyramidal cells, Neuron, 32 (2001), 141–149. |
| [2] | O. Avila-Akerberg and M. J. Chacron, Nonrenewal spike train statistics: causes and functional consequences on neural coding, Exp. Brain Res., 210 (2011), 353–371. |
| [3] | F. Zeldenrust, W. J. Wadman and B. Englitz, Neural coding with bursts–current state and future perspectives, Front. Comput. Neurosci., 12 (2018), 48. |
| [4] | A. Kepecs and J. Lisman, Information encoding and computation with spikes and bursts, Network-Comp. Neural, 14 (2003), 103–118. |
| [5] | D. A. Butts, P. O. Kanold and C. J. Shatz, A burst-based "hebbian" learning rule at retinogeniculate synapses links retinal waves to activity-dependent refinement, PLoS Biol., 5 (2007), e61. |
| [6] | J. Cocatre-Zilgien and F. Delcomyn, Identification of bursts in spike trains, J. Neurosci. Method., 41 (1992), 19–30. |
| [7] | D. C. Tam, An alternate burst analysis for detecting intra-burst firings based on inter-burst periods, Neurocomputing, 44 (2002), 1155–1159. |
| [8] | M. Chiappalone, A. Novellino, I. Vajda, et al., Burst detection algorithms for the analysis of spatio-temporal patterns in cortical networks of neurons, Neurocomputing, 65 (2005), 653–662. |
| [9] | B. Gourévitch and J. J. Eggermont, A nonparametric approach for detection of bursts in spike trains, J. Neurosci. Method., 160 (2007), 349–358. |
| [10] | L. Chen, Y. Deng, W. Luo, et al., Detection of bursts in neuronal spike trains by the mean inter-spike interval method, Prog. Nat. Sci., 19 (2009), 229–235. |
| [11] | V. Pasquale, S. Martinoia and M. Chiappalone, A self-adapting approach for the detection of bursts and network bursts in neuronal cultures, J. Comput. Neurosci., 29 (2010), 213–229. |
| [12] | D. Ko, C. Wilson, C. Lobb, et al., Detection of bursts and pauses in spike trains, J. Neurosci. Method., 211 (2012), 145–158. |
| [13] | I. A. Välkki, K. Lenk, J. E. Mikkonen, et al., Network-wide adaptive burst detection depicts neuronal activity with improved accuracy, Front. Comput. Neurosci., 11 (2017), 40. |
| [14] | D. J. Bakkum, M. Radivojevic, U. Frey, et al., Parameters for burst detection, Front. Comput. Neurosci., 7 (2014), 193. |
| [15] | C. R. Legendy and M. Salcman, Bursts and recurrences of bursts in the spike trains of sponta-neously active striate cortex neurons, J. Neurophysiol., 53 (1985), 926–939. |
| [16] | G. Palm, Evidence, information and surprise, Biol. Cybern., 42 (1981), 57–68. |
| [17] | S. Grün and S. Rotter (eds.), Analysis of Parallel Spike Trains, Springer, 2010. |
| [18] | S. Grün, M. Diesmann and A. Aertsen, 'Unitary Events' in multiple single-neuron spiking activity. I. Detection and significance, Neural Comput., 14 (2002), 43–80. |
| [19] | S. Grün, M. Diesmann and A. Aertsen, 'Unitary Events' in multiple single-neuron spiking activity. II. Non-Stationary data, Neural Comput., 14 (2002), 81–119. |
| [20] | E. Torre, D. Picado-Mui˜ no, M. Denker, et al., Statistical evaluation of synchronous spike patterns extracted by frequent item set mining, Front. Comput. Neurosci., 7 (2013), 132, |
| [21] | P. Quaglio, A. Yegenoglu, E. Torre, et al., Detection and evaluation of spatio-temporal spike patterns in massively parallel spike train data with spade, Front. Comput. Neurosci., 11 (2017), 41. |
| [22] | G. Palm, Novelty, Information and Surprise, Springer Science & Business Media, 2012. |
| [23] | M. R. DeWeese, M. Wehr and A. M. Zador, Binary spiking in auditory cortex, J. Neurosci., 23 (2003), 7940–7949. |
| [24] | H. Câteau and A. Reyes, Relation between single neuron and population spiking statistics and effects on network activity, Phys. Rev. Lett., 96 (2006), 058101. |
| [25] | T. Brochier, L. Zehl, Y. Hao, et al., Massively parallel recordings in macaque motor cortex during an instructed delayed reach-to-grasp task, Scientific Data, 5 (2018), 180055, |
| [26] | S. Tokdar, P. Xi, R. C. Kelly, et al., Detection of bursts in extracellular spike trains using hidden semi-markov point process models, J. Comput. Neurosci., 29 (2010), 203–212. |
| [27] | A. Riehle, S. Wirtssohn, S. Grün, et al., Mapping the spatio-temporal structure of motor cortical LFP and spiking activities during reach-to-grasp movements, Front. Neural Circuit., 7 (2013), 48, |
| [28] | E. Torre, P. Quaglio, M. Denker, et al., Synchronous spike patterns in macaque motor cortex during an instructed-delay reach-to-grasp task, J. Neurosci., 36 (2016), 8329–8340, |
| [29] | A. Riehle, T. Brochier, M. Nawrot, et al., Behavioral context determines network state and vari-ability dynamics in monkey motor cortex, Front. Neural Circuit., 12 (2018), 52. |
| [30] | M. P. Nawrot, C. Boucsein, V. Rodriguez Molina, et al., Measurement of variability dynamics in cortical spike trains, J. Neurosci. Method., 169 (2008), 374–390. |
| [31] | M. P. Nawrot, Analysis and interpretation of interval and count variability in neural spike trains, in Analysis of Parallel Spike Trains (eds. S. Rotter and S. Grün), Springer, Berlin, 2010. |
| [32] | C. R. Legendy, Three principles of brain function and structure, Int. J. Neurosci., 6 (1975), 237–254. |
| [33] | S. Shinomoto, H. Kim, T. Shimokawa, et al., Relating neuronal firing patterns to functional differ-entiation of cerebral cortex, PLOS Comput. Biol., 5 (2009), e1000433. |
| [34] | Y. Mochizuki, T. Onaga, H. Shimazaki, et al., Similarity in neuronal firing regimes across mam-malian species, J. Neurosci., 36 (2016), 5736–5747. |
| [35] | G. Maimon and J. A. Assad, Beyond poisson: Increased spike-time regularity across primate parietal cortex, Neuron, 62 (2009), 426–440. |
| [36] | F. Farkhooi, M. F. Strube-Bloss and M. P. Nawrot, Serial correlation in neural spike trains: Ex-perimental evidence, stochastic modeling, and single neuron variability, Phys. Rev. E, 79 (2009), 021905. |
| [37] | J. Csicsvari, H. Hirase, A. Czurko, et al., Reliability and state dependence of pyramidal cell–interneuron synapses in the hippocampus: an ensemble approach in the behaving rat, Neuron, 21 (1998), 179–189. |
| [38] | S. N. Baker and G. L. Gerstein, Determination of response latency and its application to normal-ization of cross-correlation measures, Neural Comput., 13 (2001), 1351–1377. |
| [39] | M. Nawrot, A. Aertsen and S. Rotter, Elimination of response latency variability in neuronal spike trains, Biol. Cybern., 5 (2003), 321–334. |
| [40] | A. Bollimunta, K. H. Knuth and M. Ding, Trial-by-trial estimation of amplitude and latency vari-ability in neuronal spike trains, J. Neurosci. Method., 160 (2007), 163–170. |
| [41] | M. Messer, M. Kirchner, J. Schiemann, et al., A multiple filter test for the detection of rate changes in renewal processes with varying variance, Ann. Appl. Stat., 8 (2014), 2027–2067. |
| [42] | M. Levakova, M. Tamborrino, S. Ditlevsen, et al., A review of the methods for neuronal response latency estimation, Biosystems, 136 (2015), 23–34. |
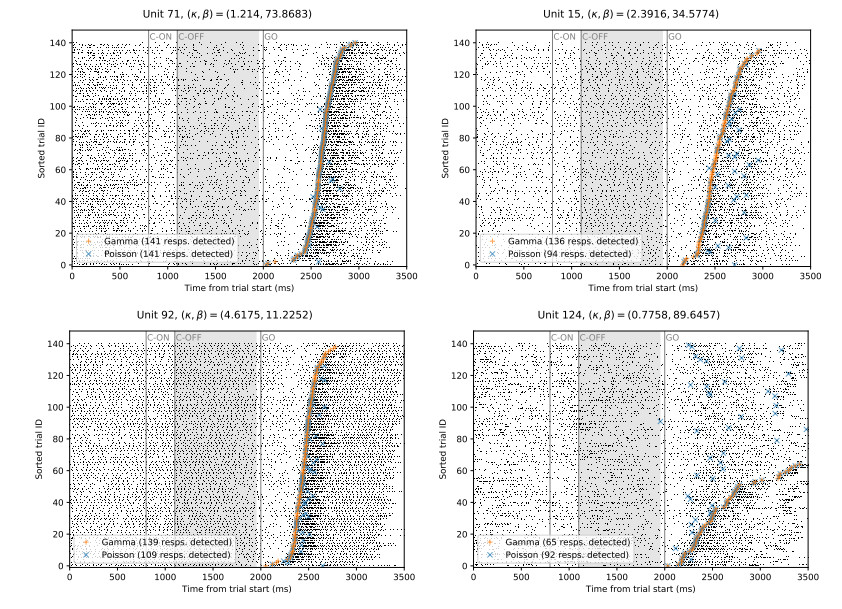
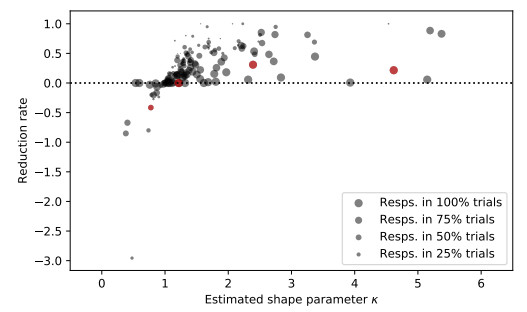
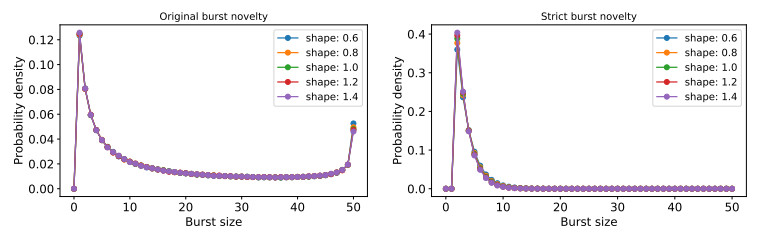
Figures(7)

Junji Ito, Emanuele Lucrezia, Günther Palm, Sonja Grün. Detection and evaluation of bursts in terms of novelty and surprise[J]. Mathematical Biosciences and Engineering, 2019, 16(6): 6990-7008. doi: 10.3934/mbe.2019351







 DownLoad:
DownLoad: