Citation: Cameron Meaney, Gibin G Powathil, Ala Yaromina, Ludwig J Dubois, Philippe Lambin, Mohammad Kohandel. Role of hypoxia-activated prodrugs in combination with radiation therapy: An in silico approach[J]. Mathematical Biosciences and Engineering, 2019, 16(6): 6257-6273. doi: 10.3934/mbe.2019312

| [1] | R. K. Carmeliet and P. Jain, Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases, Nat. Rev. Drug Discovery , 10 (2011), 417–427. |
| [2] | M. R. Horsman, L. S. Mortensen, M. Busk, et al., Imaging hypoxia to improve radiotherapy outcome, Nat. Rev. Clin. Oncol, 9 (2012), 674–687. |
| [3] | A. Vaupel and P. Mayer, Hypoxia in cancer: Significance and impact on clinical outcome, Cancer Metastasis Rev., 26 (2007), 225–239. |
| [4] | P. Hockel and M. Vaupel, Tumor Hypoxia: Definitions and Current Clinical, Biologic, and Molecular Aspects, JNCI, J. Natl. Cancer Inst., 93 (2001), 266–276. |
| [5] | K. R. Luoto, R. Kumareswaran and R. G. Bristow, Tumor hypoxia as a driving force in genetic instability, Genome Integr., 4 (2013), 5. |
| [6] | L. Harrison, Hypoxia and Anemia: Factors in Decreased Sensitivity to Radiation Therapy and Chemotherapy? Oncologist , 9 (2004), 31–40. |
| [7] | I. F. Minchinton and A. I. Tannock, Drug Penetration in Solid Tumours, Nat. Rev. Cancer, 6 (2006), 583–592. |
| [8] | I. N. Mistry, M. Thomas, E. D. D. Calder, et al., Clinical Advances of Hypoxia-Activated Prodrugs in Combination With Radiation Therapy, Int. J. Radiat. Oncol. Biol. Phys., 98 (2017), 1183–1196. |
| [9] | F. W. Hunter, B. G. Wouters and W. R. Wilson, Hypoxia-activated prodrugs: Paths forward in the era of personalised medicine, Br. J. Cancer, 114 (2016), 1071–1077. |
| [10] | J. D. Sun, Q. Liu, J. Wang, et al., TH-302, a hypoxia-activated prodrug with broad in vivo preclinical combination therapy efficacy: Optimization of dosing regimens and schedules, Cancer Chemother. Pharmacol., 69 (2012), 1487–1498. |
| [11] | S. G. Peeters, C. M. Zegers, R. Biemans, et al., TH-302 in Combination with Radiotherapy Enhances the Therapeutic Outcome and Is Associated with Pretreatment [18 F]HX4 Hypoxia PET Imaging, Clin. Cancer Res., 21 (2015), 2984–2992. |
| [12] | A. Yaromina, M. Granzier, R. Biemans, et al., A novel concept for tumour targeting with radiation: Inverse dose-painting or targeting the Low Drug Uptake Volume, Radiother. Oncology, 124 (2017), 513–520. |
| [13] | R. M. Phillips, Targeting the hypoxic fraction of tumours using hypoxiaactivated prodrugs, Cancer Chemother. Pharmacol., 77 (2016), 441–457. |
| [14] | M. Kohandel, M. Kardar, M. Milosevic, et al., Dynamics of tumor growth and combination of anti-angiogenic and cytotoxic therapies, Phys. Med. Biol., 52 (2007), 3665–3677. |
| [15] | S. F. Petit, A. L. Dekker, R. Seigneuric, et al., Intra-voxel heterogeneity influences the dose prescription for dose-painting with radiotherapy: A modelling study, Phys. Med. Biol., 54 (2009), 2179–2196. |
| [16] | G. Powathil, M. Kohandel, M. Milosevic, et al., Modeling the spatial distribution of chronic tumor hypoxia: Implications for experimental and clinical studies, Comput. Math. Methods Med., 2012 (2012), 410602. |
| [17] | S. Yonucu, Y. Defne, C. Phipps, et al., Quantifying the effects of antiangiogenic and chemotherapy drug combinations on drug delivery and treatment efficacy, PLoS Comput. Biol., 13 (2017), e1005724. |
| [18] | A. Foehrenbacher, K. Patel, M. R. Abbattista, et al., The Role of Bystander Effects in the Antitumor Activity of the Hypoxia-Activated Prodrug PR-104, Front. Oncology, 3 (2013), 1–18. |
| [19] | A. Foehrenbacher, T. W. Secomb, W. R. Wilson, et al., Design of Optimized Hypoxia-Activated Prodrugs Using Pharmacokinetic/Pharmacodynamic Modeling, Front. Oncology, 3 (2013), 33–35. |
| [20] | K. O. Hicks, F. B. Pruijn, T. W. Secomb, et al., Use of three-dimensional tissue cultures to model extravascular transport and predict in vivo activity of hypoxia-targeted anticancer drugs, J. Nat. Cancer Inst., 98 (2006), 1118–1128. |
| [21] | D. Lindsay, C. M. Garvey, S. M. Mumenthaler, et al., Leveraging Hypoxia-Activated Prodrugs to Prevent Drug Resistance in Solid Tumors, PLoS Comp. Biol., 12 (2016), 1–25. |
| [22] | T. W. Secomb, R. Hsu, R. D. Braun, et al., Theoretical simulation of oxygen transport to tumors by three-dimensional networks of microvessels, Oxygen Transp. Tissue XX, 454 (1998), 629–634. 1998. |
| [23] | R. Weinberg, The Biology of Cancer, United States: Garland Science, 2006. |
| [24] | C. Hajj, J. Russell, C. P. Hart, et al., A combination of radiation and the hypoxia-activated prodrug evofosfamide (TH-302) is efficacious against a human orthotopic pancreatic tumor model, Trans. Oncology, 10 (2017), 760–765. |
| [25] | J. K. Saggar and I. F. Tannock, Activity of the hypoxia-activated pro-drug TH-302 in hypoxic and perivascular regions of solid tumors and its potential to enhance therapeutic effects of chemotherapy, Int. J. Cancer, 134 (2014), 2726–2734. |
| [26] | G. J. Weiss, J. R. Infante, E. G. Chiorean, et al., Phase 1 study of the safety, tolerability, and pharmacokinetics of TH-302, a hypoxia-activated prodrug, in patients with advanced solid malignancies, Clin. Cancer Res., 17 (2011), 2997–3004. |
| [27] | W. R. Wilson and M. P. Hay, Targeting hypoxia in cancer therapy, Nat. Rev. Cancer, 11 (2011), 393–410. |
| [28] | G. D. Smith, Numerical solution of partial differential equations: Finite difference methods. Oxford: Oxford university press, 1985. |
| [29] | K. J. Nytko, I. Grgic, S. Bender, et al., The hypoxia-activated prodrug evofosfamide in combination with multiple regimens of radiotherapy, Oncotarget, 8 (2017), 23702–23712. |
| [30] | C. V. M. Verhagen, D. M. Vossen, K. Borgmann, et al., Fanconi anemia and homologous recombination gene variants are associated with functional DNA repair defects in vitro and poor outcome in patients with advanced head and neck squamous cell carcinoma, Oncotarget, 9 (2018), 18198–18213. |
| [31] | J. Stingele, R. Bellelli and S. J. Boulton, Mechanisms of DNA-protein crosslink repair, Nat. Rev. Mol. Cell Biol., 18 (2017), 563–573. |
| [32] | S. M. Jamieson, P. Tsai, M. K. Kondratyev, et al., Evofosfamide for the treatment of human papillomavirus-negative head and neck squamous cell carcinoma., JCI Insight, 3 (2018), e122204. |
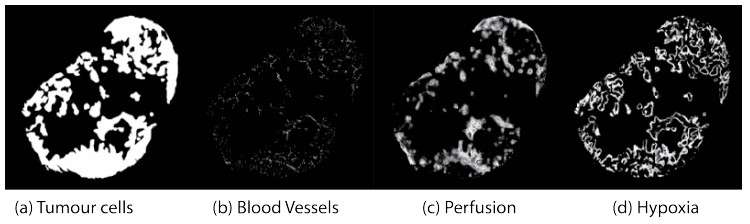
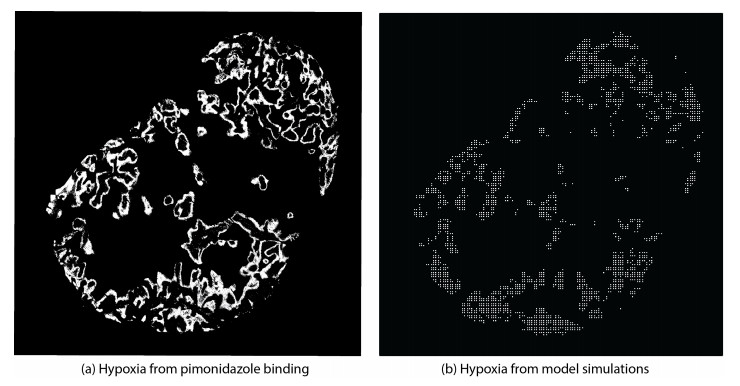
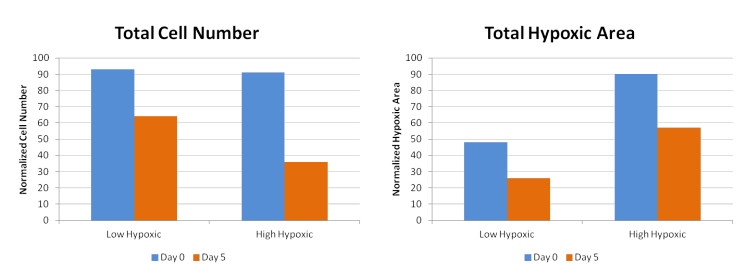
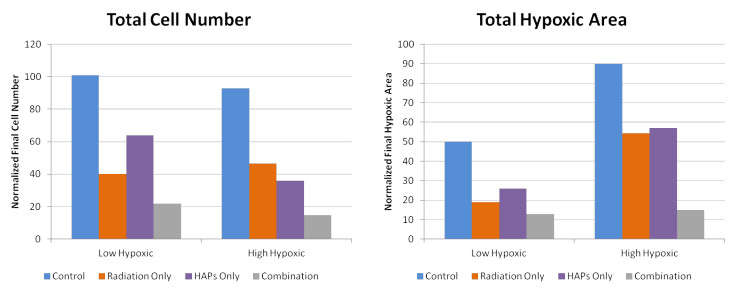
Figures(6) / Tables(1)

Cameron Meaney, Gibin G Powathil, Ala Yaromina, Ludwig J Dubois, Philippe Lambin, Mohammad Kohandel. Role of hypoxia-activated prodrugs in combination with radiation therapy: An in silico approach[J]. Mathematical Biosciences and Engineering, 2019, 16(6): 6257-6273. doi: 10.3934/mbe.2019312







 DownLoad:
DownLoad: