Citation: Yahia Swilem, Hanan AL-Otaibi. Structural studies of nucleation and growth of Cu and Fe nanoparticles using XAFS simulation[J]. AIMS Materials Science, 2020, 7(1): 1-8. doi: 10.3934/matersci.2020.1.1

| [1] |
Shipway AN, Katz E, Willner I (2000) Nanoparticle arrays on surfaces for electronic, optical, and sensor applications. ChemPhysChem 1: 18-52. doi: 10.1002/1439-7641(20000804)1:1<18::AID-CPHC18>3.0.CO;2-L

|
| [2] | Amos PI, Louis H, Adegoke KA, et al. (2018) Understanding the mechanism of electrochemical reduction of CO2 using Cu/Cu-based electrodes: A review. Asian J Nanosci Mater 1: 183-224. |
| [3] |
Athanasiou EK, Grass RN, Stark WJ (2006) Large-scale production of carbon-coated copper nanoparticles for sensor applications. Nanotechnology 17: 1668-1673. doi: 10.1088/0957-4484/17/6/022
|
| [4] |
Laaksonen A, Talanquer V, Oxtoby DW (1995) Nucleation: measurements, theory, and atmospheric applications. Annu Rev Phys Chem 46: 489-524. doi: 10.1146/annurev.pc.46.100195.002421
|
| [5] |
Rehr JJ, Kas JJ, Vila FD, et al. (2010) Parameter-free calculations of X-ray spectra with FEFF9. Phys Chem Chem Phys 12: 5503-5513. doi: 10.1039/b926434e
|
| [6] | Mathew K, Zheng C, Winston D, et al. (2018) High-throughput computational X-ray absorption spectroscopy. Figshare https://doi.org/10.6084/m9.figshare.c.3946561. |
| [7] | Wyckoff RWG (1963) Crystal Structures, 2 Eds., New York: John Wiley and Sons, 1: 7-83. |
| [8] |
Swilem Y (2013) EXAFS studies of nanostructured finemet-type alloys. Cryst Res Technol 48: 374-380. doi: 10.1002/crat.201300039
|
| [9] |
Swilem Y, Sobczak E, Nietubyć R, et al. (2005) EXAFS analysis of nanocrystallization process in Fe85Zr7B6Cu2 alloys by using cumulant method. Physica B 364: 71-77. doi: 10.1016/j.physb.2005.03.037
|
| [10] |
Ravel B, Newville M (2005) ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J Synchrotron Rad 12: 537-541. doi: 10.1107/S0909049505012719
|
| [11] |
Bazin D, Rehr JJ (2003) Limits and advantages of X-ray absorption near edge structure for nanometer scale metallic clusters. J Phys Chm B 107: 12398-12402. doi: 10.1021/jp0223051
|
| [12] | Lalena JN, Cleary DA (2010) Principles of Inorganic Mmaterials Design, 2 Eds., Hoboken: John Wiley and Sons, 1: 109-132. |
| [13] |
Hahn JE, Scott RA, Hodgson KO, et al. (1982) Observation of an electric quadrupole transition in the X-ray absorption spectrum of a Cu(II) complex. Chem Phys Lett 88: 595-598. doi: 10.1016/0009-2614(82)85016-1
|
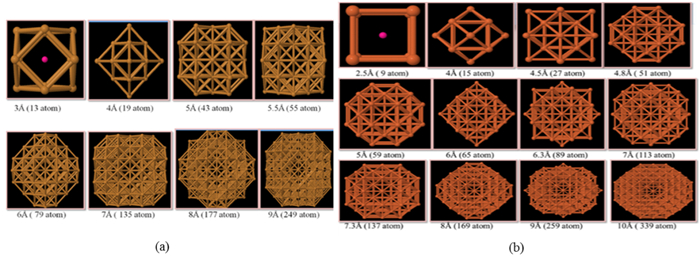
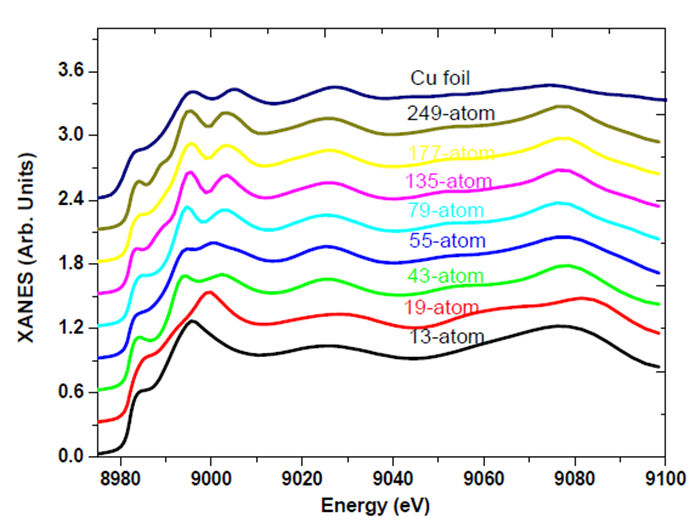
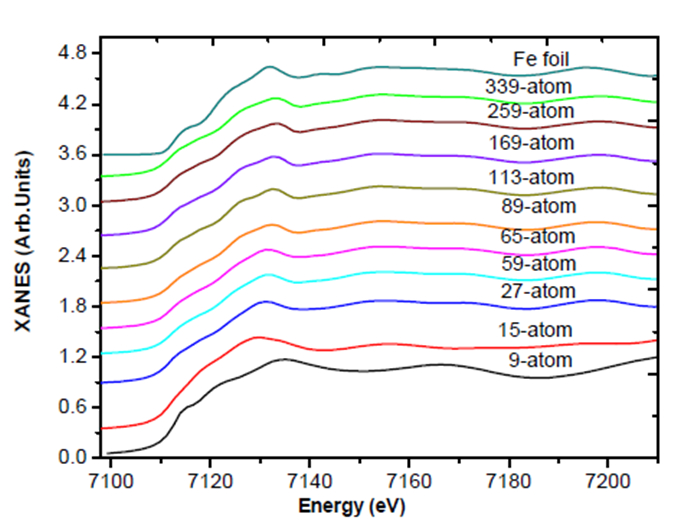
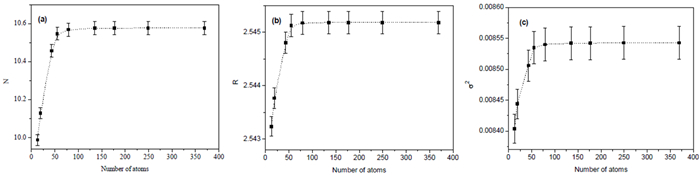
Figures(7)

Yahia Swilem, Hanan AL-Otaibi. Structural studies of nucleation and growth of Cu and Fe nanoparticles using XAFS simulation[J]. AIMS Materials Science, 2020, 7(1): 1-8. doi: 10.3934/matersci.2020.1.1







 DownLoad:
DownLoad: