Citation: Agni Hadjilouka, Konstantinos Nikolidakis, Spiros Paramithiotis, Eleftherios H. Drosinos. Effect of co-culture with enterocinogenic E. faecium on L. monocytogenes key virulence gene expression[J]. AIMS Microbiology, 2016, 2(3): 359-371. doi: 10.3934/microbiol.2016.3.359

| [1] | Baek SY, Lim SY, Lee DH, et al. (2000) Incidence and characterization of Listeria monocytogenes from domestic and imported foods in Korea. J Food Prot 63: 186–189. |
| [2] |
Cordano AM, Rocourt J (2001) Occurrence of Listeria monocytogenes in food in Chile. Int J Food Microbiol 70: 175–178. doi: 10.1016/S0168-1605(01)00533-5

|
| [3] |
Fallah AA, Saei-Dehkordi SS, Rahnama M, et al. (2012) Prevalence and antimicrobial resistance patterns of Listeria species isolated from poultry products marketed in Iran. Food Control 28: 327–332. doi: 10.1016/j.foodcont.2012.05.014
|
| [4] |
Wang G, Qian W, Zhang X, et al. (2015) Prevalence, genetic diversity and antimicrobial resistance of Listeria monocytogenes isolated from ready-to-eat meat products in Nanjing, China. Food Control 50: 202–208. doi: 10.1016/j.foodcont.2014.07.057
|
| [5] | Khen BK, Lynch OA, Carroll J, et al. (2015) Occurrence, antibiotic resistance and molecular characterization of Listeria monocytogenes in the beef chain in the Republic of Ireland. Zoonoses Public Health 62: 11–17. |
| [6] | Montero D, Bodero M, Riveros G, et al. (2015) Molecular epidemiology and genetic diversity of Listeria monocytogenes isolates from a wide variety of ready-to-eat foods and their relationship to clinical strains from listeriosis outbreaks in Chile. Front Microbiol 6: 384. |
| [7] | Jami M, Ghanbari M, Zunabovic M, et al. (2014) Listeria monocytogenes in aquatic food products—a review. Comprehensive Reviews in Food Science & Food Safety 13: 798–813. |
| [8] | Lin CM, Fernando SY, Wei CI (1996) Occurrence of Listeria monocytogenes, Salmonella spp., Escherichia coli and E. coli O157:H7 in vegetable salads. Food Control 7: 135–40. |
| [9] | Sado PN, Jinneman KC, Husby GJ, et al. (1998) Identification of Listeria monocytogenes from unpasteurized apple juice using rapid test kits. J Food Prot 61: 1199–1202. |
| [10] |
Francis GA, Thomas C, O'Beirne D (1999) The microbiological safety of minimally processed vegetables. Int J Food Sci Tech 34: 1–22. doi: 10.1046/j.1365-2621.1999.00253.x
|
| [11] | Abadias M, Usall J, Anguera M, et al. (2008) Microbiological quality of fresh, minimally-processed fruits and vegetables, and sprouts from retail establishments. Int J Food Microbiol 123: 121–129. |
| [12] |
Cordano AM, Jacquet C (2009) Listeria monocytogenes isolated from vegetable salads sold at supermarkets in Santiago, Chile: Prevalence and strain characterization. Int J Food Microbiol 132: 176–179. doi: 10.1016/j.ijfoodmicro.2009.04.008
|
| [13] |
Kramarenko T, Roasto M, Meremäe K, et al. (2013) Listeria monocytogenes prevalence and serotype diversity in various foods. Food Control 30: 24–29. doi: 10.1016/j.foodcont.2012.06.047
|
| [14] | Roche SM, Velge P, Liu D (2008) Virulence determination, In: Liu D (Ed.) Hanbook of Listeria monocytogenes. CRC Press, 241–270. |
| [15] |
Dussurget O (2008) New insights into determinants of Listeria monocytogenes virulence. Int Rev Cell Mol Biol 270: 1–38. doi: 10.1016/S1937-6448(08)01401-9
|
| [16] |
Bohne J, Sokolovic Z, Goebel W (1994) Transcriptional regulation of prfA and PrfA-regulated virulence genes in Listeria monocytogenes. Mol Microbiol 11: 1141–1150. doi: 10.1111/j.1365-2958.1994.tb00390.x
|
| [17] |
Renzoni A, Cossart P, Dramsi S (1999) PrfA, the transcriptional activator of virulence genes, is upregulated during interaction of Listeria monocytogenes with mammalian cells and in eukaryotic cell extracts. Mol Microbiol 34: 552–561. doi: 10.1046/j.1365-2958.1999.01621.x
|
| [18] |
Nadon CA, Bowen BM, Wiedmann M, et al. (2002) Sigma B contributes to PrfA-mediated virulence in Listeria monocytogenes. Infect Immun 70: 3948–3952. doi: 10.1128/IAI.70.7.3948-3952.2002
|
| [19] | Schluter D, Domann E, Buck C, et al. (1998) Phosphatidylcholine-specific phospholipase C from Listeria monocytogenes is an important virulence factor in murine cerebral listeriosis. Infect Immun 66: 5930–5938. |
| [20] |
Grundling A, Gonzalez MD, Higgins DE (2003) Requirement of the Listeria monocytogenes broad-range phospholipase PC-PLC during infection of human epithelial cells. J Bacteriol 185: 6295–6307. doi: 10.1128/JB.185.21.6295-6307.2003
|
| [21] | Domann E, Leimeister-Wachter M, Goebel W, et al. (1991) Molecular cloning, sequencing, and identification of a metalloprotease gene from Listeria monocytogenes that is species specific and physically linked to the listeriolysin gene. Infect Immun 59: 65–72. |
| [22] | Raveneau J, Geoffroy C, Beretti JL, et al. (1992) Reduced virulence of a Listeria monocytogenes phospholipase-deficient mutant obtained by transposon insertion into the zinc metalloprotease gene. Infect Immun 60: 916–921. |
| [23] |
Suarez M, Gonzalez-Zorn B, Vega Y, et al. (2001) A role for ActA in epithelial cell invasion by Listeria monocytogenes. Cell Microbiol 3: 853–864. doi: 10.1046/j.1462-5822.2001.00160.x
|
| [24] |
Birmingham CL, Canadien V, Gouin E, et al. (2007) Listeria monocytogenes evades killing by autophagy during colonization of host cells. Autophagy 3: 442–451. doi: 10.4161/auto.4450
|
| [25] |
Travier L, Guadagnini S, Gouin E, et al. (2013) Acta promotes Listeria monocytogenes aggregation, intestinal colonization and carriage. PLoS Pathog 9: e1003131. doi: 10.1371/journal.ppat.1003131
|
| [26] |
Bierne H, Sabet C, Personnic N, et al. (2007) Internalins: a complex family of leucine-rich repeat-containing proteins in Listeria monocytogenes. Microbes Infect 9: 1156–1166. doi: 10.1016/j.micinf.2007.05.003
|
| [27] |
Liu D, Lawrence ML, Ainsworth AJ, et al. (2007) Toward an improved laboratory definition of Listeria monocytogenes virulence. Int J Food Microbiol 118: 101–115. doi: 10.1016/j.ijfoodmicro.2007.07.045
|
| [28] | Devriese LA, Pot B (1995) The genus Enterococcus, In: Wood BJB, Holzapfel WH, The Lactic Acid Bacteria, Eds., London: Springer US, 327–367. |
| [29] |
Hazards EPOB (2013) Scientific opinion on the maintenance of the list of QPS biological agents intentionally added to food and feed (2013 update). EFSA Journal 11: 3449. doi: 10.2903/j.efsa.2013.3449
|
| [30] |
Moreno MRF, Rea MC, Cogan TM, et al. (2003) Applicability of a bacteriocin-producing Enterococcus faecium as a co-culture in Cheddar cheese manufacture. Int J Food Microbiol 81: 73– 84. doi: 10.1016/S0168-1605(02)00167-8
|
| [31] | Achemchem F, Abrini J, Martínez-Bueno M, et al. (2006) Control of Listeria monocytogenes in goat's milk and goat's Jben by the bacteriocinogenic Enterococcus faecium F58 strain. J Food Prot 69: 2370–2376. |
| [32] |
Rubio R, Bover-Cid S, Martin B, et al. (2013) Assessment of safe enterococci as bioprotective cultures in low-acid fermented sausages combined with high hydrostatic pressure. Food Microbiol 33: 158–165. doi: 10.1016/j.fm.2012.09.012
|
| [33] |
Liu W, Zhang L, Shi J, et al. (2015) Assessment of the safety and applications of bacteriocinogenic Enterococcus faecium Y31 as an adjunct culture in North-eastern Chinese traditional fermentation paocai. Food Control 50: 637–644. doi: 10.1016/j.foodcont.2014.10.004
|
| [34] |
Garriga M, Rubio R, Aymerich T, et al. (2015) Potentially probiotic and bioprotective lactic acid bacteria starter cultures antagonise the Listeria monocytogenes adhesion to HT29 colonocyte-like cells. Benef Microbes 6: 337–343. doi: 10.3920/BM2014.0056
|
| [35] | Leimeister-Wachter M, Domann E, Chakraborty T (1992) The expression of virulence genes in Listeria monocytogenes is thermoregulated. J Bacteriol 174: 947–952. |
| [36] |
Milenbachs Lukowiak A, Mueller KJ, Freitag NE, et al. (2004) Deregulation of Listeria monocytogenes virulence gene expression by two distinct and semi-independent pathways. Microbiol 150: 321–333. doi: 10.1099/mic.0.26718-0
|
| [37] |
Milenbachs AA, Brown DP, Moors M, et al. (1997) Carbon-source regulation of virulence gene expression in Listeria monocytogenes. Mol Microbiol 23: 1075–1085. doi: 10.1046/j.1365-2958.1997.2711634.x
|
| [38] | Renzoni A, Klarsfeld A, Dramsi S, et al. (1997) Evidence that PrfA, the pleiotropic activator of virulence genes in Listeria monocytogenes, can be present but inactive. Infect Immun 65: 1515–1518. |
| [39] | Behari J, Youngman P (1998) Regulation of hly expression in Listeria monocytogenes by carbon sources and pH occurs through separate mechanisms mediated by PrfA. Infect Immun 66, 3635–3642. |
| [40] |
Chaturongakul S, Raengpradub S, Wiedmann M, et al. (2008) Modulation of stress and virulence in Listeria monocytogenes. Trends Microbiol 16: 388–396. doi: 10.1016/j.tim.2008.05.006
|
| [41] |
Sheikh-Zeinoddin M, Perehinec TM, Hill SE, et al. (2000) Maillard reaction causes suppression of virulence gene expression in Listeria monocytogenes. Int J Food Microbiol 61: 41–49. doi: 10.1016/S0168-1605(00)00371-8
|
| [42] |
Bowman JP, Bittencourt CR, Ross T (2008) Differential gene expression of Listeria monocytogenes during high hydrostatic pressure processing. Microbiol 154: 462–475. doi: 10.1099/mic.0.2007/010314-0
|
| [43] |
Kastbjerg VG, Larsen MH, Gram L, et al. (2010) Influence of sublethal concentrations of common disinfectants on expression of virulence genes in Listeria monocytogenes. Appl Environ Microbiol 76: 303–309. doi: 10.1128/AEM.00925-09
|
| [44] |
Stasiewicz MJ, Wiedmann M, Bergholz TM (2011) The transcriptional response of Listeria monocytogenes during adaptation to growth on lactate and diacetate includes synergistic changes that increase fermentative acetoin production. Appl Environ Microbiol 77: 5294–5306. doi: 10.1128/AEM.02976-10
|
| [45] |
Shi H, Trinh Q, Xu W, et al. (2013) The transcriptional response of virulence genes in Listeria monocytogenes during inactivation by nisin. Food Control 31: 519–524. doi: 10.1016/j.foodcont.2012.11.008
|
| [46] |
Duodu S, Holst-Jensen A, Skjerdal T, et al. (2010) Influence of storage temperature on gene expression and virulence potential of Listeria monocytogenes strains grown in a salmon matrix. Food Microbiol 27: 795–801. doi: 10.1016/j.fm.2010.04.012
|
| [47] |
Olesen I, Thorsen L, Jespersen L (2010) Relative transcription of Listeria monocytogenes virulence genes in liver pates with varying NaCl content. Int J Food Microbiol 141: S60–S68. doi: 10.1016/j.ijfoodmicro.2010.01.042
|
| [48] |
Rantsiou K, Greppi A, Garosi M, et al. (2012) Strain dependent expression of stress response and virulence genes of Listeria monocytogenes in meat juices as determined by microarray. Int J Food Microbiol 152: 116–122. doi: 10.1016/j.ijfoodmicro.2011.08.009
|
| [49] | Rantsiou K, Mataragas M, Alessandria V, et al. (2012) Expression of virulence genes of Listeria monocytogenes in food. J Food Saf 32: 161–168. |
| [50] |
Alessandria V, Rantsiou K, Dolci P, et al. (2013) Comparison of gene expression of Listeria monocytogenes in vitro and in the soft cheese Crescenza. Int J Dairy Technol 66: 83–89. doi: 10.1111/1471-0307.12008
|
| [51] |
Mataragas M, Rovetto F, Bellio A, et al. (2015) Differential gene expression profiling of Listeria monocytogenes in Cacciatore and Felino salami to reveal potential stress resistance biomarkers. Food Microbiol 46: 408–417. doi: 10.1016/j.fm.2014.09.003
|
| [52] | Hadjilouka A, Molfeta C, Panagiotopoulou O, et al. (2016) Expression of Listeria monocytogenes key virulence genes during growth in liquid medium, on rocket and melon at 4, 10 and 30 °C. Food Microbiol 55: 7–15. |
| [53] | Paramithiotis S, Vlontartzik E, Drosinos EH (2014) Enterocin production by Enterococcus faecium strains isolated from Greek spontaneously fermented sausages. Ital J Food Sci 26: 12–17. |
| [54] |
Andersen CL, Jensen JL, Orntoft TF (2004) Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res 64: 5245–5250. doi: 10.1158/0008-5472.CAN-04-0496
|
| [55] | Kubista M, Sindelka R, Tichopad A, et al. (2007) The Prime Technique. Real-time PCR Data Analysis. G.I.T. Lab. J. 9–10, 33–35. Available from: http://www.tataa.com/wp-content/uploads/2012/09/GIT_Laboratory-Journal_33-35_2007.pdf. (Access date: 8 October 2013). |
| [56] | Giraffa G, Carminati D (1997) Control of Listeria monocytogenes in the rind of Taleggio, a surface-smear cheese, by a bacteriocin from Enterococcus faecium 7C5. Sci Aliment 17: 383–391. |
| [57] | Izquierdo E, Marchioni E, Aoude-Werner D, et al. (2009) Smearing of soft cheese with Enterococcus faecium WHE 81, a multi-bacteriocin producer, against Listeria monocytogenes. Food Microbiol 26: 16–20. |
| [58] |
Huang Y, Ye K, Yu K, et al. (2016) The potential influence of two Enterococcus faecium on the growth of Listeria monocytogenes. Food Control 67: 18–24. doi: 10.1016/j.foodcont.2016.02.009
|
| [59] |
Berghe EVD, Winter TD, Vuyst LD (2006) Enterocin A production by Enterococcus faecium FAIR-E 406 is characterised by a temperature- and pH-dependent switch-off mechanism when growth is limited due to nutrient depletion. Int J Food Microbiol 107: 159–170. doi: 10.1016/j.ijfoodmicro.2005.08.027
|
| [60] |
Stark L, Giersch T, Wünschiers R (2014) Efficiency of RNA extraction from selected bacteria in the context of biogas production and metatranscriptomics. Anaerobe 29: 85–90. doi: 10.1016/j.anaerobe.2013.09.007
|
| [61] | Becker LA, Cetin MS, Hutkins RW, et al. (1998) Identification of the gene encoding the alternative sigma factor B from Listeria monocytogenes and its role in osmotolerance. J Bacteriol 180: 4547–4554. |
| [62] |
Ferreira A, O'Byrne CP, Boor KJ (2001) Role of sigma B in heat, ethanol, acid, and oxidative stress resistance and during carbon starvation in Listeria monocytogenes. Appl Environ Microbiol 67: 4454–4457. doi: 10.1128/AEM.67.10.4454-4457.2001
|
| [63] |
Ferreira A, Sue D, O'Byrne CP, et al. (2003) Role of Listeria monocytogenes sigma B in survival of lethal acidic conditions and in the acquired acid tolerance response. Appl Environ Microbiol 69: 2692–2698. doi: 10.1128/AEM.69.5.2692-2698.2003
|
| [64] | Wiedmann M, Arvik TJ, Hurley RJ, et al. (1998) General stress transcription factor sigma B and its role in acid tolerance and virulence of Listeria monocytogenes. J Bacteriol 180: 3650–3656. |
| [65] |
Chan YC, Boor KJ, Wiedmann M (2007) sigma B -dependent and sigma B -independent mechanisms contribute to transcription of Listeria monocytogenes cold stress genes during cold shock and cold growth. Appl Environ Microbiol 73: 6019–6029. doi: 10.1128/AEM.00714-07
|
| [66] |
Veen SVD, Abee T (2010) Importance of SigB for Listeria monocytogenes static and continuous-flow biofilm formation and disinfectant resistance. Appl Environ Microbiol 76: 7854–7860. doi: 10.1128/AEM.01519-10
|
| [67] |
Port GC, Freitag NE (2007) Identification of novel Listeria monocytogenes secreted virulence factors following mutational activation of the central virulence regulator, PrfA. Infect Immun 75: 5886–5897. doi: 10.1128/IAI.00845-07
|
| [68] |
Greene SL, Freitag NE (2003) Negative regulation of PrfA, the key activator of Listeria monocytogenes virulence gene expression, is dispensable for bacterial pathogenesis. Microbiol 149: 111–120. doi: 10.1099/mic.0.25692-0
|
| [69] |
Ollinger J, Bowen B, Wiedmann M, et al. (2009) Listeria monocytogenes sigma B modulates PrfA-mediated virulence factor expression. Infect Immun 77: 2113–2124. doi: 10.1128/IAI.01205-08
|
| [70] |
Kazmierczak MJ, Wiedmann M, Boor KJ (2006) Contributions of Listeria monocytogenes sigma B and PrfA to expression of virulence and stress response genes during extra- and intracellular growth. Microbiol 152: 1827–1838. doi: 10.1099/mic.0.28758-0
|
| [71] |
Williams JR, Thayyullathil C, Freitag NE (2000) Sequence variations within PrfA DNA binding sites and effects on Listeria monocytogenes virulence gene expression. J Bacteriol 182: 837–841. doi: 10.1128/JB.182.3.837-841.2000
|
| [72] |
McGann P, Raengpradub S, Ivanek R, et al. (2008) Differential regulation of Listeria monocytogenes internalin and internalin-like genes by sigma B and PrfA as revealed by subgenomic microarray analyses. Foodborne Pathog Dis 5: 417–435. doi: 10.1089/fpd.2008.0085
|
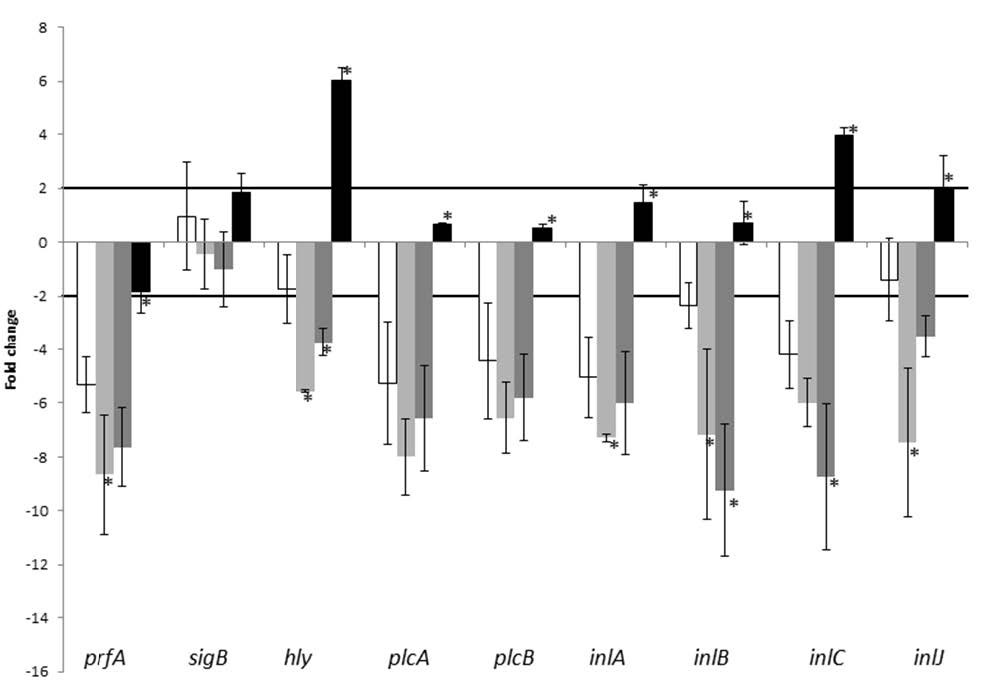
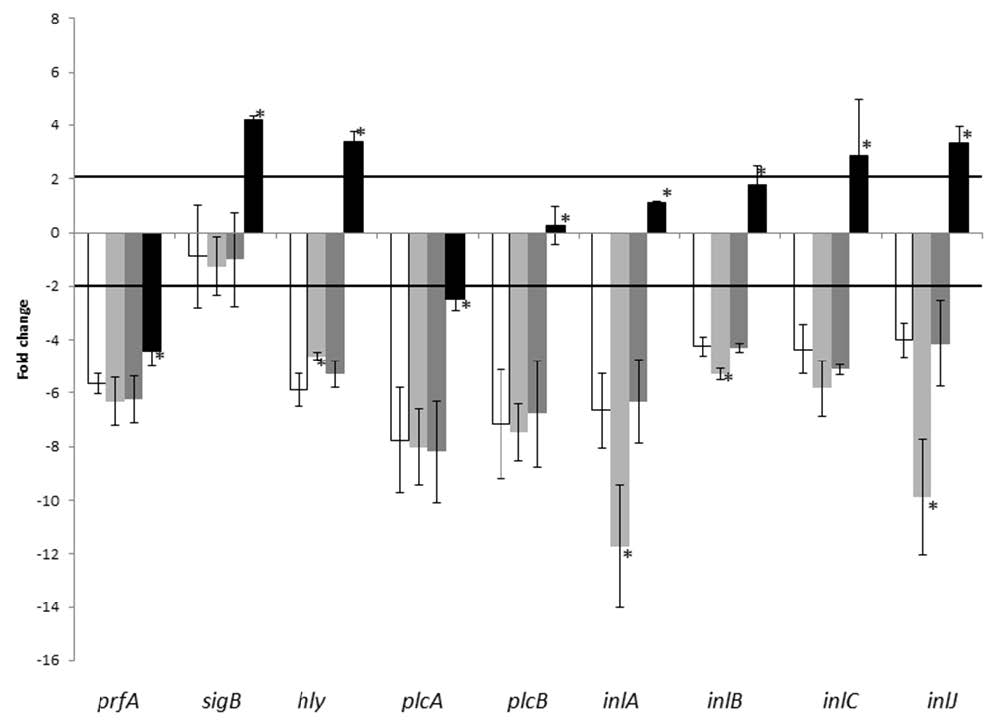
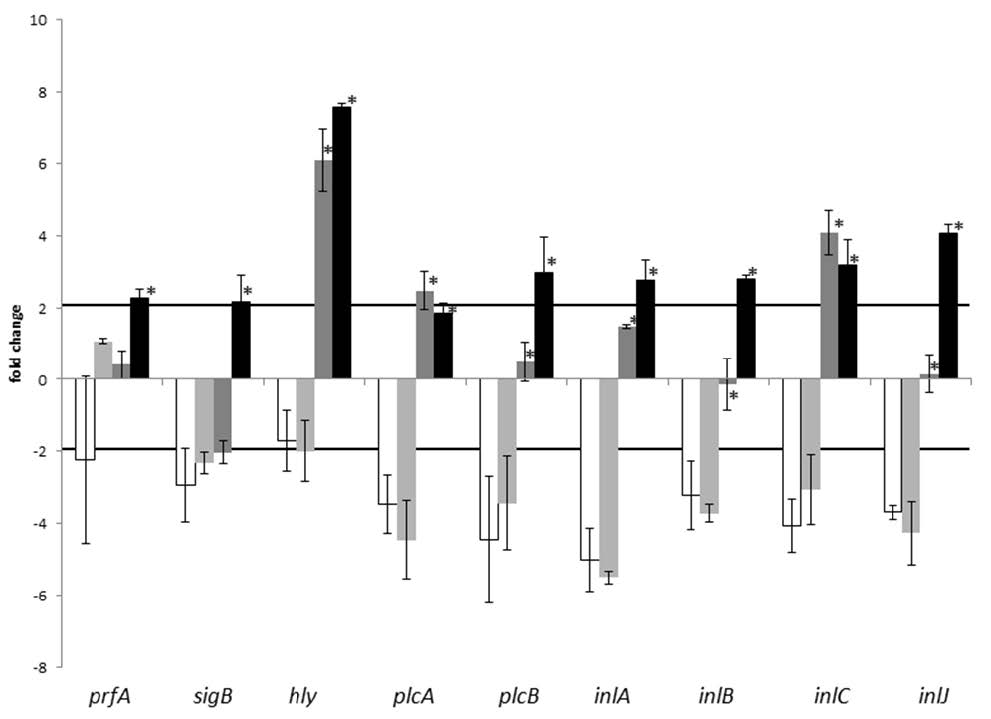
Figures(3) / Tables(2)

Agni Hadjilouka, Konstantinos Nikolidakis, Spiros Paramithiotis, Eleftherios H. Drosinos. Effect of co-culture with enterocinogenic E. faecium on L. monocytogenes key virulence gene expression[J]. AIMS Microbiology, 2016, 2(3): 359-371. doi: 10.3934/microbiol.2016.3.359







 DownLoad:
DownLoad: