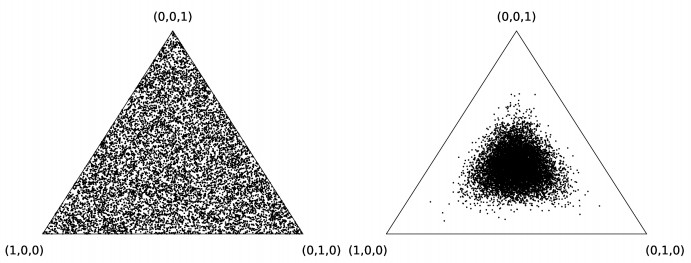
Bayesian Network (BN) modeling is a prominent and increasingly popular computational systems biology method. It aims to construct network graphs from the large heterogeneous biological datasets that reflect the underlying biological relationships. Currently, a variety of strategies exist for evaluating BN methodology performance, ranging from utilizing artificial benchmark datasets and models, to specialized biological benchmark datasets, to simulation studies that generate synthetic data from predefined network models. The last is arguably the most comprehensive approach; however, existing implementations often rely on explicit and implicit assumptions that may be unrealistic in a typical biological data analysis scenario, or are poorly equipped for automated arbitrary model generation. In this study, we develop a purely probabilistic simulation framework that addresses the demands of statistically sound simulations studies in an unbiased fashion. Additionally, we expand on our current understanding of the theoretical notions of causality and dependence / conditional independence in BNs and the Markov Blankets within.
Citation: Grigoriy Gogoshin, Sergio Branciamore, Andrei S. Rodin. Synthetic data generation with probabilistic Bayesian Networks[J]. Mathematical Biosciences and Engineering, 2021, 18(6): 8603-8621. doi: 10.3934/mbe.2021426
Bayesian Network (BN) modeling is a prominent and increasingly popular computational systems biology method. It aims to construct network graphs from the large heterogeneous biological datasets that reflect the underlying biological relationships. Currently, a variety of strategies exist for evaluating BN methodology performance, ranging from utilizing artificial benchmark datasets and models, to specialized biological benchmark datasets, to simulation studies that generate synthetic data from predefined network models. The last is arguably the most comprehensive approach; however, existing implementations often rely on explicit and implicit assumptions that may be unrealistic in a typical biological data analysis scenario, or are poorly equipped for automated arbitrary model generation. In this study, we develop a purely probabilistic simulation framework that addresses the demands of statistically sound simulations studies in an unbiased fashion. Additionally, we expand on our current understanding of the theoretical notions of causality and dependence / conditional independence in BNs and the Markov Blankets within.

| [1] | S. Branciamore, G. Gogoshin, M. Di Giulio, A. S. Rodin, Intrinsic properties of TRNA molecules as deciphered via bayesian network and distribution divergence analysis, Life (Basel), 8 (2018), E5. |
| [2] |
X. Zhang, S. Branciamore, G. Gogoshin, A. S. Rodin, Analysis of high-resolution 3d intrachromosomal interactions aided by bayesian network modeling, Proc. Natl. Acad. Sci. USA, 114 (2017), E10359–E10368. doi: 10.1073/pnas.1620425114

|
| [3] |
A. S. Rodin, G. Gogoshin, S. Hilliard, L. Wang, C. Egelston, R. C. Rockne, et al., Dissecting response to cancer immunotherapy by applying bayesian network analysis to flow cytometry data, Int. J. Mol. Sci., 22 (2021), 2316. doi: 10.3390/ijms22052316
|
| [4] |
A. J. Sedgewick, K. Buschur, I. Shi, J. D. Ramsey, V. K. Raghu, D. V. Manatakis, et al., Mixed graphical models for integrative causal analysis with application to chronic lung disease diagnosis and prognosis, Bioinformatics, 35 (2019), 1204–1212. doi: 10.1093/bioinformatics/bty769
|
| [5] | A. K. Becker, M. Dörr, S. B. Felix, F. Frost, H. J. Grabe, M. M. Lerch, et al., From heterogeneous healthcare data to disease-specific biomarker networks: A hierarchical bayesian network approach, PLoS Comput. Biol., 17 (2021). |
| [6] |
G. Gogoshin, E. Boerwinkle, A. S. Rodin, New algorithm and software (bnomics) for inferring and visualizing bayesian networks from heterogeneous "big" biological and genetic data, J. Comput. Biol., 24 (2017), 340–356. doi: 10.1089/cmb.2016.0100
|
| [7] |
A. Rodin, A. Brown, A. G. Clark, C. F. Sing, E. Boerwinkle, Mining genetic epidemiology data with bayesian networks: Application to apoe gene variants and plasma lipid levels, J. Comput. Biol., 12 (2005), 1–11. doi: 10.1089/cmb.2005.12.1
|
| [8] | F. F. Sherif, N. Zayed, M. Fakhr, Discovering alzheimer genetic biomarkers using bayesian networks, Adv. Bioinform., 2015 (2015), 639367. |
| [9] |
L. Wang, P. Audenaert, T. Michoel, High-dimensional bayesian network inference from systems genetics data using genetic node ordering, Front. Genet., 10 (2019), 1196. doi: 10.3389/fgene.2019.01196
|
| [10] | Z. Lan, Y. Zhao, J. Kang, T. Yu, Bayesian network feature finder (banff): an r package for gene network feature selection, Bioinformatics, 32 (2016), 3685–3687. |
| [11] | R. Neapolitan, D. Xue, X. Jiang, Modeling the altered expression levels of genes on signaling pathways in tumors as causal bayesian networks, Cancer Inform., 13 (2014), 77–84. |
| [12] | A. van de Stolpe, W. Verhaegh, J.-Y. Blay, C. X. Ma, P. Pauwels, M. Pegram, et al., RNA based approaches to profile oncogenic pathways from low quantity samples to drive precision oncology strategies, Front. Genet., 11 (2021). |
| [13] | Q. Qi, J. Li, J. Cheng, Reconstruction of metabolic pathways by combining probabilistic graphical model-based and knowledge-based methods, BMC Proc., 8 (2014), S5. |
| [14] | D. Pe'er, Bayesian network analysis of signaling networks: a primer, Sci. Signal., 2005 (2005), pl4. |
| [15] | G. Piatetsky-Shapiro, P. Tamayo, Microarray data mining: facing the challenges, SIGKDD Explor. Newsl., 5 (2003), 1-5. |
| [16] |
Z. Zeng, X. Jiang, R. Neapolitan, Discovering causal interactions using bayesian network scoring and information gain, BMC Bioinform., 17 (2016), 221. doi: 10.1186/s12859-016-1084-8
|
| [17] |
J. D. Ziebarth, A. Bhattacharya, Y. Cui, Bayesian network webserver: a comprehensive tool for biological network modeling, Bioinformatics, 29 (2013), 2801–3. doi: 10.1093/bioinformatics/btt472
|
| [18] | Q. Zhang, X. Shi, A mixture copula bayesian network model for multimodal genomic data, Cancer Inform., 16 (2017). |
| [19] | Y. Zhao, C. Chang, M. Hannum, J. Lee, R. Shen, Bayesian network-driven clustering analysis with feature selection for high-dimensional multi-modal molecular data, Sci. Rep., 11 (2021). |
| [20] | J. Pearl, Probabilistic reasoning in intelligent systems, 1988. |
| [21] | J. Pearl, Causality, Cambridge Univ. Press, 2009. |
| [22] | S. Russell, P. Norvig, Artificial intelligence: A modern approach, 3rd edition, Prentice Hall, 2010. |
| [23] | P. Spirtes, C. Glymour, R. Scheines, Causation, prediction, and search, 2nd edition, MIT Press, 2000. |
| [24] |
C. Glymour, K. Zhang, P. Spirtes, Review of causal discovery methods based on graphical models, Front. Genet., 10 (2019), 524. doi: 10.3389/fgene.2019.00524
|
| [25] | D. Heckerman, D. Geiger, D. Chickering, Learning bayesian networks: The combination of knowledge and statistical data, Mach. Learn., 20 (1995), 197–243. |
| [26] |
P. Spirtes, K. Zhang, Causal discovery and inference: concepts and recent methodological advances, Appl. Inform. (Berl)., 3 (2016), 3. doi: 10.1186/s40535-016-0018-x
|
| [27] |
K. Zhang, B. Schölkopf, P. Spirtes, C. Glymour, Learning causality and causality-related learning: some recent progress, Natl. Sci. Rev., 5 (2018), 26–29. doi: 10.1093/nsr/nwx137
|
| [28] |
V. K. Raghu, J. D. Ramsey, A. Morris, D. V. Manatakis, P. Sprites, P. K. Chrysanthis, et al., Comparison of strategies for scalable causal discovery of latent variable models from mixed data, Int. J. Data Sci. Anal., 6 (2018), 33–45. doi: 10.1007/s41060-018-0104-3
|
| [29] |
J. Ramsey, M. Glymour, R. Sanchez-Romero, C. Glymour, A million variables and more: the fast greedy equivalence search algorithm for learning high-dimensional graphical causal models, with an application to functional magnetic resonance images, Int. J. Data Sci. Anal., 3 (2017), 121–129. doi: 10.1007/s41060-016-0032-z
|
| [30] |
L. Xing, M. Guo, X. Liu, C. Wang, L. Wang, Y. Zhang, An improved bayesian network method for reconstructing gene regulatory network based on candidate auto selection, BMC Genom., 18 (2017), 844. doi: 10.1186/s12864-017-4228-y
|
| [31] |
L. Zhang, L. O. Rodrigues, N. R. Narain, V. R. Akmaev, bAIcis: A novel bayesian network structural learning algorithm and its comprehensive performance evaluation against open-source software, J. Comput. Biol., 27 (2020), 698–708. doi: 10.1089/cmb.2019.0210
|
| [32] |
B. Andrews, J. Ramsey, G. F. Cooper, Scoring bayesian networks of mixed variables, Int. J. Data Sci. Anal., 6 (2018), 3–18. doi: 10.1007/s41060-017-0085-7
|
| [33] | B. Andrews, J. Ramsey, G. F. Cooper, Learning high-dimensional directed acyclic graphs with mixed data-types, Proc. Mach. Learn. Res., 104 (2019), 4–21. |
| [34] |
A. J. Sedgewick, I. Shi, R. M. Donovan, P. V. Benos, Learning mixed graphical models with separate sparsity parameters and stability-based model selection, BMC Bioinform., 17 (2016), 175. doi: 10.1186/s12859-016-1039-0
|
| [35] |
F. Jabbari, J. Ramsey, P. Spirtes, G. Cooper, Discovery of causal models that contain latent variables through bayesian scoring of independence constraints, Lect. Notes Comput. Sc., 10535 (2017), 142–157. doi: 10.1007/978-3-319-71246-8_9
|
| [36] | J. M. Ogarrio, P. Spirtes, R. J, A hybrid causal search algorithm for latent variable models, JMLR Workshop Conf. Proc., 52 (2016), 368–379. |
| [37] | K. Yu, L. Liu, J. Li, Learning markov blankets from multiple interventional data sets, IEEE Trans. Neural Netw. Learn. Syst., 31 (2020). |
| [38] |
J. Chen, R. Zhang, X. Dong, L. Lin, Y. Zhu, J. He, et al., shinybn: an online application for interactive bayesian network inference and visualization, BMC Bioinform., 20 (2019), 711. doi: 10.1186/s12859-019-3309-0
|
| [39] |
T. Eicher, A. Patt, E. Kautto, R. Machiraju, E. Mathé, Y. Zhang, Challenges in proteogenomics: a comparison of analysis methods with the case study of the dream proteogenomics sub-challenge, BMC Bioinform., 20 (2019), 669. doi: 10.1186/s12859-019-3253-z
|
| [40] |
N. Ramanan, S. Natarajan, Causal learning from predictive modeling for observational data, Front. Big Data, 3 (2020), 535976. doi: 10.3389/fdata.2020.535976
|
| [41] |
S. Tasaki, B. Sauerwine, B. Hoff, H. Toyoshiba, C. Gaiteri, E. C. Neto, Bayesian network reconstruction using systems genetics data: comparison of mcmc methods, Genetics, 199 (2015), 973–89. doi: 10.1534/genetics.114.172619
|
| [42] | A. Pratapa, A. P. Jalihal, J. N. Law, A. Bharadwaj, T. M. Murali, Benchmarking algorithms for gene regulatory network inference from single-cell transcriptomic data, Nat. Methods, 17 (2020). |
| [43] | J. Peters, J. M. Mooij, D. Janzing, B. Schölkopf, Causal discovery with continuous additive noise models, J. Mach. Learn. Res., 15 (2014), 2009–2053, |
| [44] | D. Kaur, M. Sobiesk, S. Patil, J. Liu, P. Bhagat, A. Gupta, et al., Application of bayesian networks to generate synthetic health data, J. Am. Med. Inform. Assoc., 28 (2020), 801–811. |
| [45] | J. B. Young, P. Graham, R. Penny, Using bayesian networks to create synthetic data, Qual. Eng., 55 (2010), 363–366. |
| [46] |
R. Roozegar, A. R. Soltani, On the asymptotic behavior of randomly weighted averages, Stat. Probabil. Lett., 96 (2015), 269–272. doi: 10.1016/j.spl.2014.10.003
|


Figures(3)

Grigoriy Gogoshin, Sergio Branciamore, Andrei S. Rodin. Synthetic data generation with probabilistic Bayesian Networks[J]. Mathematical Biosciences and Engineering, 2021, 18(6): 8603-8621. doi: 10.3934/mbe.2021426







 DownLoad:
DownLoad: