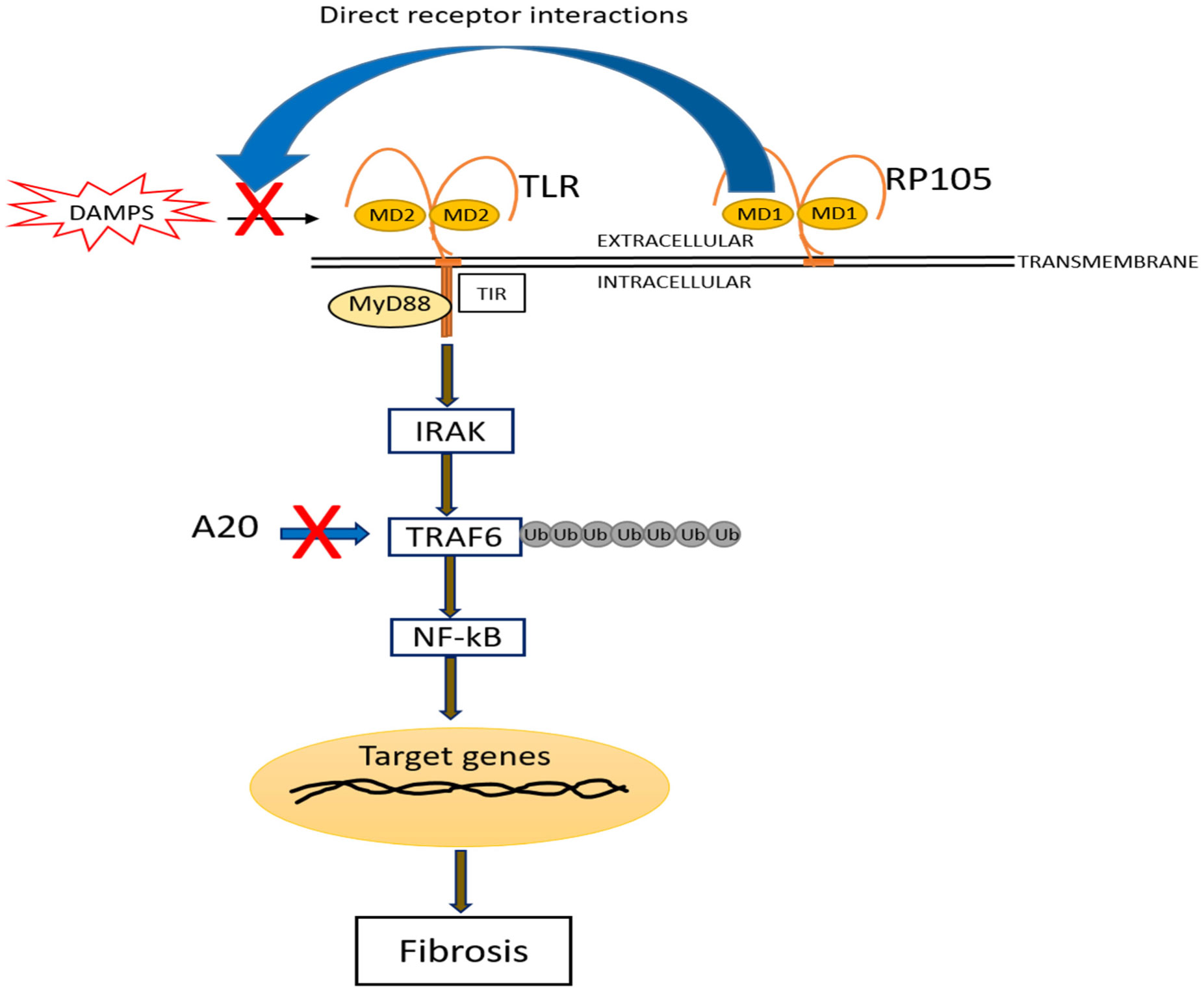
Toll-like receptors (TLRs) are essential defensive mediators implicated in immune diseases. Tight regulation of TLR function is indispensable to avoid the damaging effects of chronic signaling. Several endogenous molecules have emerged as negative regulators of TLR signaling. In this review, we highlighted the structure, regulation, and function of RP105 and A20 in negatively modulating TLR-dependent inflammatory diseases, and in fibrosis and potential therapeutic approaches.
Citation: Swarna Bale, John Varga, Swati Bhattacharyya. Role of RP105 and A20 in negative regulation of toll-like receptor activity in fibrosis: potential targets for therapeutic intervention[J]. AIMS Allergy and Immunology, 2021, 5(2): 102-126. doi: 10.3934/Allergy.2021009
Toll-like receptors (TLRs) are essential defensive mediators implicated in immune diseases. Tight regulation of TLR function is indispensable to avoid the damaging effects of chronic signaling. Several endogenous molecules have emerged as negative regulators of TLR signaling. In this review, we highlighted the structure, regulation, and function of RP105 and A20 in negatively modulating TLR-dependent inflammatory diseases, and in fibrosis and potential therapeutic approaches.

| [1] |
Medzhitov R, Preston-Hurlburt P, Janeway CA (1997) A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 388: 394-397. doi: 10.1038/41131

|
| [2] |
Stoffels JMJ, Zhao C, Baron W (2013) Fibronectin in tissue regeneration: timely disassembly of the scaffold is necessary to complete the build. Cell Mol Life Sci 70: 4243-4253. doi: 10.1007/s00018-013-1350-0
|
| [3] | Bhattacharyya S, Wang W, Morales-Nebreda L, et al. (2016) Tenascin-C drives persistence of organ fibrosis. Nat Commun 7: 1-14. |
| [4] |
Huang QQ, Sobkoviak R, Jockheck-Clark AR, et al. (2009) Heat shock protein 96 is elevated in rheumatoid arthritis and activates macrophages primarily via TLR2 signaling. J Immunol 182: 4965-4973. doi: 10.4049/jimmunol.0801563
|
| [5] |
Huang QQ, Pope RM (2010) Toll-like receptor signaling: a potential link among rheumatoid arthritis, systemic lupus, and atherosclerosis. J Leukocyte Biol 88: 253-262. doi: 10.1189/jlb.0310126
|
| [6] |
Vencken SF, Greene CM (2016) Toll-like receptors in cystic fibrosis: impact of dysfunctional microRNA on innate immune responses in the cystic fibrosis lung. J Innate Immun 8: 541-549. doi: 10.1159/000444687
|
| [7] |
Bhattacharyya S, Wang W, Qin W, et al. (2018) TLR4-dependent fibroblast activation drives persistent organ fibrosis in skin and lung. JCI Insight 3: e98850. doi: 10.1172/jci.insight.98850
|
| [8] |
Divanovic S, Trompette A, Atabani SF, et al. (2005) Inhibition of TLR-4/MD-2 signaling by RP105/MD-1. J Endotoxin Res 11: 363-368. doi: 10.1177/09680519050110061201
|
| [9] |
Gon Y, Asai Y, Hashimoto S, et al. (2004) A20 inhibits toll-like receptor 2- and 4-mediated interleukin-8 synthesis in airway epithelial cells. Am J Resp Cell Mol 31: 330-336. doi: 10.1165/rcmb.2003-0438OC
|
| [10] |
Peng J, Tao X, Li R, et al. (2015) Novel toll/IL-1 receptor homologous region adaptors act as negative regulators in amphioxus TLR signaling. J Immunol 195: 3110-3118. doi: 10.4049/jimmunol.1403003
|
| [11] |
Divanovic S, Trompette A, Petiniot LK, et al. (2007) Regulation of TLR4 signaling and the host interface with pathogens and danger: the role of RP105. J Leukocyte Biol 82: 265-271. doi: 10.1189/jlb.0107021
|
| [12] |
Zhao X, Chu Q, Cui J, et al. (2018) MicroRNA-19a as a negative regulator in TLR signaling pathway by direct targeting myeloid differentiation factor 88 in miiuy croaker. Dev Comp Immunol 87: 171-175. doi: 10.1016/j.dci.2018.06.009
|
| [13] |
Xu M, Li D, Yang C, et al. (2018) MicroRNA-34a inhibition of the TLR signaling pathway via CXCL10 suppresses breast cancer cell invasion and migration. Cell Physiol Biochem 46: 1286-1304. doi: 10.1159/000489111
|
| [14] | Xie J, Zhang L, Fan X, et al. (2019) MicroRNA-146a improves sepsis-induced cardiomyopathy by regulating the TLR-4/NF-κB signaling pathway. Exp Ther Med 18: 779-785. |
| [15] |
Curtale G, Mirolo M, Renzi TA, et al. (2013) Negative regulation of Toll-like receptor 4 signaling by IL-10-dependent microRNA-146b. P Natl Acad Sci USA 110: 11499-11504. doi: 10.1073/pnas.1219852110
|
| [16] |
Jung WJ, Lee SY, Choi SI, et al. (2018) Toll-like receptor expression in pulmonary sensory neurons in the bleomycin-induced fibrosis model. PLoS One 13: e0193117. doi: 10.1371/journal.pone.0193117
|
| [17] |
Stärkel P, Schnabl B, Leclercq S, et al. (2019) Deficient IL-6/stat3 signaling, high TLR7, and type I interferons in early human alcoholic liver disease: A triad for liver damage and fibrosis. Hepatol Commun 3: 867-882. doi: 10.1002/hep4.1364
|
| [18] |
Castellano G, Stasi A, Franzin R, et al. (2019) LPS-binding protein modulates acute renal fibrosis by inducing pericyte-to-myofibroblast trans-differentiation through TLR-4 signaling. Int J Mol Sci 20: 3682. doi: 10.3390/ijms20153682
|
| [19] |
Cáceres FT, Gaspari TA, Samuel CS, et al. (2019) Serelaxin inhibits the profibrotic TGF-β1/IL-1β axis by targeting TLR-4 and the NLRP3 inflammasome in cardiac myofibroblasts. FASEB J 33: 14717-14733. doi: 10.1096/fj.201901079RR
|
| [20] | Liu AJ, Wu PC, Ciou JR, et al. (2021) Differential expression of Toll-like receptors 1 and 3 in patients with systemic lupus erythematosus and systemic sclerosis. Research Square In press. |
| [21] |
Bhattacharyya S, Varga J (2015) Emerging roles of innate immune signaling and toll-like receptors in fibrosis and systemic sclerosis. Curr Rheumatol Rep 17: 2. doi: 10.1007/s11926-014-0474-z
|
| [22] |
Beutler B (2009) Microbe sensing, positive feedback loops, and the pathogenesis of inflammatory diseases. Immunol Rev 227: 248-263. doi: 10.1111/j.1600-065X.2008.00733.x
|
| [23] |
West AP, Koblansky AA, Ghosh S (2006) Recognition and signaling by toll-like receptors. Annu Rev Cell Dev Biol 22: 409-437. doi: 10.1146/annurev.cellbio.21.122303.115827
|
| [24] |
Kawai T, Akira S (2011) Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34: 637-650. doi: 10.1016/j.immuni.2011.05.006
|
| [25] |
Kumar H, Kawai T, Akira S (2011) Pathogen recognition by the innate immune system. Int Rev Immunol 30: 16-34. doi: 10.3109/08830185.2010.529976
|
| [26] |
Howell J, Angus P, Gow P, et al. (2013) Toll-like receptors in hepatitis C infection: Implications for pathogenesis and treatment. J Gastroen Hepatol 28: 766-776. doi: 10.1111/jgh.12170
|
| [27] |
Liu G, Zhao Y (2007) Toll-like receptors and immune regulation: their direct and indirect modulation on regulatory CD4+ CD25+ T cells. Immunology 122: 149-156. doi: 10.1111/j.1365-2567.2007.02651.x
|
| [28] |
Yao C, Oh JH, Lee DH, et al. (2015) Toll-like receptor family members in skin fibroblasts are functional and have a higher expression compared to skin keratinocytes. Int J Mol Med 35: 1443-1450. doi: 10.3892/ijmm.2015.2146
|
| [29] |
Price AE, Shamardani K, Lugo KA, et al. (2018) A map of toll-like receptor expression in the intestinal epithelium reveals distinct spatial, cell type-specific, and temporal patterns. Immunity 49: 560-575. doi: 10.1016/j.immuni.2018.07.016
|
| [30] |
Yamamoto M, Sato S, Hemmi H, et al. (2003) TRAM is specifically involved in the Toll-like receptor 4-mediated MyD88-independent signaling pathway. Nat Immunol 4: 1144-1150. doi: 10.1038/ni986
|
| [31] |
Yamamoto M, Sato S, Hemmi H, et al. (2003) Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 301: 640-643. doi: 10.1126/science.1087262
|
| [32] |
Meier A, Alter G, Frahm N, et al. (2007) MyD88-dependent immune activation mediated by human immunodeficiency virus type 1-encoded Toll-like receptor ligands. J Virol 81: 8180-8191. doi: 10.1128/JVI.00421-07
|
| [33] |
Cronin JG, Turner ML, Goetze L, et al. (2012) Toll-like receptor 4 and MYD88-dependent signaling mechanisms of the innate immune system are essential for the response to lipopolysaccharide by epithelial and stromal cells of the bovine endometrium. Biol Reprod 86: 51. doi: 10.1095/biolreprod.111.092718
|
| [34] |
Kawagoe T, Sato S, Matsushita K, et al. (2008) Sequential control of Toll-like receptor–dependent responses by IRAK1 and IRAK2. Nat Immunol 9: 684. doi: 10.1038/ni.1606
|
| [35] |
Kawai T, Akira S (2007) Signaling to NF-κB by Toll-like receptors. Trends Mol Med 13: 460-469. doi: 10.1016/j.molmed.2007.09.002
|
| [36] |
Akira S, Takeda K (2004) Toll-like receptor signalling. Nat Rev Immunol 4: 499-511. doi: 10.1038/nri1391
|
| [37] |
Adhikari A, Xu M, Chen ZJ (2007) Ubiquitin-mediated activation of TAK1 and IKK. Oncogene 26: 3214-3226. doi: 10.1038/sj.onc.1210413
|
| [38] |
Chen F, Bhatia D, Chang Q, et al. (2006) Finding NEMO by K63-linked polyubiquitin chain. Cell Death Differ 13: 1835. doi: 10.1038/sj.cdd.4402014
|
| [39] |
Anwar MA, Basith S, Choi S (2013) Negative regulatory approaches to the attenuation of Toll-like receptor signaling. Exp Mol Med 45: e11-e11. doi: 10.1038/emm.2013.28
|
| [40] |
Bibeau-Poirier A, Servant MJ (2008) Roles of ubiquitination in pattern-recognition receptors and type I interferon receptor signaling. Cytokine 43: 359-367. doi: 10.1016/j.cyto.2008.07.012
|
| [41] |
Wang C, Chen T, Zhang J, et al. (2009) The E3 ubiquitin ligase Nrdp1 ‘preferentially’ promotes TLR-mediated production of type I interferon. Nat Immunol 10: 744-752. doi: 10.1038/ni.1742
|
| [42] |
Wang T, Chuang TH, Ronni T, et al. (2006) Flightless I homolog negatively modulates the TLR pathway. J Immunol 176: 1355-1362. doi: 10.4049/jimmunol.176.3.1355
|
| [43] |
Chen Z, Zhou R, Zhang Y, et al. (2020) β-arrestin 2 quenches TLR signaling to facilitate the immune evasion of EPEC. Gut Microbes 11: 1423-1437. doi: 10.1080/19490976.2020.1759490
|
| [44] |
Kobayashi K, Hernandez LD, Galán JE, et al. (2002) IRAK-M is a negative regulator of Toll-like receptor signaling. Cell 110: 191-202. doi: 10.1016/S0092-8674(02)00827-9
|
| [45] |
Al-Shaghdali K, Durante B, Hayward C, et al. (2019) Macrophage subsets exhibit distinct E. coli-LPS tolerisable cytokines associated with the negative regulators, IRAK-M and Tollip. PLoS One 14: e0214681. doi: 10.1371/journal.pone.0214681
|
| [46] |
Jiang S, Li X, Hess NJ, et al. (2016) TLR10 is a negative regulator of both MyD88-dependent and-independent TLR signaling. J Immunol 196: 3834-3841. doi: 10.4049/jimmunol.1502599
|
| [47] |
Olmos-Ortiz A, Déciga-García M, Preciado-Martínez E, et al. (2019) Prolactin decreases LPS-induced inflammatory cytokines by inhibiting TLR-4/NFκB signaling in the human placenta. Mol Hum Reprod 25: 660-667. doi: 10.1093/molehr/gaz038
|
| [48] |
Cao Y, Sun Y, Chang H, et al. (2019) The E3 ubiquitin ligase RNF 182 inhibits TLR-triggered cytokine production through promoting p65 ubiquitination and degradation. FEBS Lett 593: 3210-3219. doi: 10.1002/1873-3468.13583
|
| [49] |
Song Y, Li P, Qin L, et al. (2021) CUL4B negatively regulates Toll-like receptor-triggered proinflammatory responses by repressing Pten transcription. Cell Mol Immunol 18: 339-349. doi: 10.1038/s41423-019-0323-0
|
| [50] |
Lou Y, Han M, Liu H, et al. (2020) Essential roles of S100A10 in Toll-like receptor signaling and immunity to infection. Cell Mol Immunol 17: 1053-1062. doi: 10.1038/s41423-019-0278-1
|
| [51] | Hu YH, Wang Y, Wang F, et al. (2020) SPOP negatively regulates Toll-like receptor-induced inflammation by disrupting MyD88 self-association. Cell Mol Immunol In press. |
| [52] |
Jiang G, Gong M, Song H, et al. (2020) Tob2 Inhibits TLR-Induced Inflammatory Responses by Association with TRAF6 and MyD88. J Immunol 205: 981-986. doi: 10.4049/jimmunol.2000057
|
| [53] | Miyake K, Yamashita Y, Ogata M, et al. (1995) RP105, a novel B cell surface molecule implicated in B cell activation, is a member of the leucine-rich repeat protein family. J Immunol 154: 3333-3340. |
| [54] |
Divanovic S, Trompette A, Atabani SF, et al. (2005) Negative regulation of Toll-like receptor 4 signaling by the Toll-like receptor homolog RP105. Nat Immunol 6: 571-578. doi: 10.1038/ni1198
|
| [55] |
Kimoto M, Nagasawa K, Miyake K (2003) Role of TLR4/MD-2 and RP105/MD-1 in innate recognition of lipopolysaccharide. Scand J Infect Dis 35: 568-572. doi: 10.1080/00365540310015700
|
| [56] |
Ogata H, Su I, Miyake K, et al. (2000) The toll-like receptor protein RP105 regulates lipopolysaccharide signaling in B cells. J Exp Med 192: 23-30. doi: 10.1084/jem.192.1.23
|
| [57] |
Nagai Y, Shimazu R, Ogata H, et al. (2002) Requirement for MD-1 in cell surface expression of RP105/CD180 and B-cell responsiveness to lipopolysaccharide. Blood 99: 1699-1705. doi: 10.1182/blood.V99.5.1699
|
| [58] |
Miyake K, Yamashita Y, Hitoshi Y, et al. (1994) Murine B cell proliferation and protection from apoptosis with an antibody against a 105-kD molecule: unresponsiveness of X-linked immunodeficient B cells. J Exp Med 180: 1217-1224. doi: 10.1084/jem.180.4.1217
|
| [59] |
Chan VWF, Mecklenbräuker I, Su I, et al. (1998) The molecular mechanism of B cell activation by toll-like receptor protein RP-105. J Exp Med 188: 93-101. doi: 10.1084/jem.188.1.93
|
| [60] |
Grumont RJ, Rourke IJ, O'Reilly LA, et al. (1998) B lymphocytes differentially use the Rel and nuclear factor κB1 (NF-κB1) transcription factors to regulate cell cycle progression and apoptosis in quiescent and mitogen-activated cells. J Exp Med 187: 663-674. doi: 10.1084/jem.187.5.663
|
| [61] |
Lardenoye JHP, Delsing DJM, De Vries MR, et al. (2000) Accelerated atherosclerosis by placement of a perivascular cuff and a cholesterol-rich diet in ApoE*3Leiden transgenic mice. Circ Res 87: 248-253. doi: 10.1161/01.RES.87.3.248
|
| [62] |
Allen JL, Flick LM, Divanovic S, et al. (2012) Cutting edge: regulation of TLR4-driven B cell proliferation by RP105 is not B cell autonomous. J Immunol 188: 2065-2069. doi: 10.4049/jimmunol.1103282
|
| [63] |
Karper JC, Ewing MM, de Vries MR, et al. (2013) TLR accessory molecule RP105 (CD180) is involved in post-interventional vascular remodeling and soluble RP105 modulates neointima formation. PLoS One 8: e67923. doi: 10.1371/journal.pone.0067923
|
| [64] |
Karper JC, de Jager SCA, Ewing MM, et al. (2013) An unexpected intriguing effect of Toll-like receptor regulator RP105 (CD180) on atherosclerosis formation with alterations on B-cell activation. Arterioscl Throm Vas 33: 2810-2817. doi: 10.1161/ATVBAHA.113.301882
|
| [65] |
Wezel A, van der Velden D, Maassen JM, et al. (2015) RP105 deficiency attenuates early atherosclerosis via decreased monocyte influx in a CCR2 dependent manner. Atherosclerosis 238: 132-139. doi: 10.1016/j.atherosclerosis.2014.11.020
|
| [66] |
Wezel A, De Vries MR, Maassen JM, et al. (2016) Deficiency of the TLR4 analogue RP105 aggravates vein graft disease by inducing a pro-inflammatory response. Sci Rep 6: 1-13. doi: 10.1038/srep24248
|
| [67] |
Yang J, Zeng P, Yang J, et al. (2019) The Role of RP105 in cardiovascular disease through regulating TLR4 and PI3K signaling pathways. Curr Med Sci 39: 185-189. doi: 10.1007/s11596-019-2017-3
|
| [68] |
Yang J, Guo X, Yang J, et al. (2015) RP105 protects against apoptosis in ischemia/reperfusion-induced myocardial damage in rats by suppressing TLR4-mediated signaling pathways. Cell Physiol Biochem 36: 2137-2148. doi: 10.1159/000430180
|
| [69] |
Li X, Yang J, Yang J, et al. (2016) RP105 protects against myocardial ischemia–reperfusion injury via suppressing TLR4 signaling pathways in rat model. Exp Mol Pathol 100: 281-286. doi: 10.1016/j.yexmp.2015.12.016
|
| [70] |
Xiong X, Liu Y, Mei Y, et al. (2017) Novel protective role of myeloid differentiation 1 in pathological cardiac remodelling. Sci Rep 7: 1-13. doi: 10.1038/s41598-016-0028-x
|
| [71] |
Guo X, Jiang H, Yang J, et al. (2016) Radioprotective 105 kDa protein attenuates ischemia/reperfusion-induced myocardial apoptosis and autophagy by inhibiting the activation of the TLR4/NF-κB signaling pathway in rats. Int J Mol Med 38: 885-893. doi: 10.3892/ijmm.2016.2686
|
| [72] | Guo X, Jiang H, Chen J, et al. (2018) RP105 ameliorates hypoxia/reoxygenation injury in cardiac microvascular endothelial cells by suppressing TLR4/MAPKs/NF-κB signaling. Int J Mol Med 42: 505-513. |
| [73] |
Yang Y, Yang J, Liu X, et al. (2018) Down-regulation of miR-327 alleviates ischemia/reperfusion-induced myocardial damage by targeting RP105. Cell Physiol Biochem 49: 1090-1104. doi: 10.1159/000493288
|
| [74] |
Qin Q, Cui L, Zhou Z, et al. (2019) Inhibition of microRNA-141-3p reduces hypoxia-induced apoptosis in H9c2 rat cardiomyocytes by activating the RP105-dependent PI3K/AKT signaling pathway. Med Sci Monit 25: 7016. doi: 10.12659/MSM.916361
|
| [75] | Sun Y, Liu L, Yuan J, et al. (2018) RP105 protects PC12 cells from oxygen–glucose deprivation/reoxygenation injury via activation of the PI3K/AKT signaling pathway. Int J Mol Med 41: 3081-3089. |
| [76] |
Yu CH, Micaroni M, Puyskens A, et al. (2015) RP105 engages phosphatidylinositol 3-kinase p110δ to facilitate the trafficking and secretion of cytokines in macrophages during mycobacterial infection. J Immunol 195: 3890-3900. doi: 10.4049/jimmunol.1500017
|
| [77] |
Yazawa N, Fujimoto M, Sato S, et al. (2003) CD19 regulates innate immunity by the toll-like receptor RP105 signaling in B lymphocytes. Blood 102: 1374-1380. doi: 10.1182/blood-2002-11-3573
|
| [78] |
Honda Y, Yamagiwa S, Matsuda Y, et al. (2007) Altered expression of TLR homolog RP105 on monocytes hypersensitive to LPS in patients with primary biliary cirrhosis. J Hepatol 47: 404-411. doi: 10.1016/j.jhep.2007.03.012
|
| [79] |
Zhang Z, La Placa D, Nguyen T, et al. (2019) CEACAM1 regulates the IL-6 mediated fever response to LPS through the RP105 receptor in murine monocytes. BMC Immunol 20: 1-16. doi: 10.1186/s12865-018-0284-6
|
| [80] |
Koarada S, Tada Y, Ushiyama O, et al. (1999) B cells lacking RP105, a novel B cell antigen, in systemic lupus erythematosus. Arthritis Rheum 42: 2593-2600. doi: 10.1002/1529-0131(199912)42:12<2593::AID-ANR12>3.0.CO;2-G
|
| [81] |
Kikuchi Y, Koarada S, Tada Y, et al. (2001) Difference in B cell activation between dermatomyositis and polymyositis: analysis of the expression of RP105 on peripheral blood B cells. Ann Rheum Dis 60: 1137-1140. doi: 10.1136/ard.60.12.1137
|
| [82] |
Koarada S, Tada Y, Kikuchi Y, et al. (2001) CD180 (RP105) in rheumatic diseases. Rheumatology 40: 1315-1316. doi: 10.1093/rheumatology/40.11.1315
|
| [83] | Koarada S, Tada Y (2012) RP105-negative B cells in systemic lupus erythematosus. Clin Dev Immunol 2012: 1-5. |
| [84] | Koarada S, Tada Y, Suematsu R, et al. (2012) Phenotyping of P105-negative B cell subsets in patients with systemic lupus erythematosus. Clin Dev Immunol 2012: 1-8. |
| [85] |
Korganow AS, Knapp AM, Nehme-Schuster H, et al. (2010) Peripheral B cell abnormalities in patients with systemic lupus erythematosus in quiescent phase: decreased memory B cells and membrane CD19 expression. J Autoimmun 34: 426-434. doi: 10.1016/j.jaut.2009.11.002
|
| [86] |
Erdő-Bonyár S, Rapp J, Minier T, et al. (2019) Toll-Like receptor mediated activation of natural autoantibody producing b cell subpopulations in an autoimmune disease model. Int J Mol Sci 20: 6152. doi: 10.3390/ijms20246152
|
| [87] |
Harhaj EW, Dixit VM (2012) Regulation of NF-κB by deubiquitinases. Immunol Rev 246: 107-124. doi: 10.1111/j.1600-065X.2012.01100.x
|
| [88] |
Aksentijevich I, Zhou Q (2017) NF-κB pathway in autoinflammatory diseases: dysregulation of protein modifications by ubiquitin defines a new category of autoinflammatory diseases. Front Immunol 8: 399. doi: 10.3389/fimmu.2017.00399
|
| [89] |
Keusekotten K, Elliott PR, Glockner L, et al. (2013) OTULIN antagonizes LUBAC signaling by specifically hydrolyzing Met1-linked polyubiquitin. Cell 153: 1312-1326. doi: 10.1016/j.cell.2013.05.014
|
| [90] |
Dixit VM, Green S, Sarma V, et al. (1990) Tumor necrosis factor-alpha induction of novel gene products in human endothelial cells including a macrophage-specific chemotaxin. J Biol Chem 265: 2973-2978. doi: 10.1016/S0021-9258(19)39896-5
|
| [91] |
Kinsella S, Fichtner M, Watters O, et al. (2018) Increased A20-E3 ubiquitin ligase interactions in bid-deficient glia attenuate TLR3- and TLR4-induced inflammation. J Neuroinflammation 15: 1-12. doi: 10.1186/s12974-018-1143-3
|
| [92] |
Bhattacharyya S, Varga J (2018) Endogenous ligands of TLR4 promote unresolving tissue fibrosis: Implications for systemic sclerosis and its targeted therapy. Immunol Lett 195: 9-17. doi: 10.1016/j.imlet.2017.09.011
|
| [93] |
Feng H, Pyykkö I, Zou J (2016) Involvement of ubiquitin-editing protein A20 in modulating inflammation in rat cochlea associated with silver nanoparticle-induced CD68 upregulation and TLR4 activation. Nanoscale Res Lett 11: 1-13. doi: 10.1186/s11671-015-1209-4
|
| [94] |
Krikos A, Laherty CD, Dixit VM (1992) Transcriptional activation of the tumor necrosis factor alpha-inducible zinc finger protein, A20, is mediated by kappa B elements. J Biol Chem 267: 17971-17976. doi: 10.1016/S0021-9258(19)37138-8
|
| [95] |
Opipari AW, Boguski MS, Dixit VM (1990) The A20 cDNA induced by tumor necrosis factor alpha encodes a novel type of zinc finger protein. J Biol Chem 265: 14705-14708. doi: 10.1016/S0021-9258(18)77165-2
|
| [96] |
Catrysse L, Vereecke L, Beyaert R, et al. (2014) A20 in inflammation and autoimmunity. Trends Immunol 35: 22-31. doi: 10.1016/j.it.2013.10.005
|
| [97] |
Lee EG, Boone DL, Chai S, et al. (2000) Failure to regulate TNF-induced NF-κB and cell death responses in A20-deficient mice. Science 289: 2350-2354. doi: 10.1126/science.289.5488.2350
|
| [98] |
Lu TT, Onizawa M, Hammer GE, et al. (2013) Dimerization and ubiquitin mediated recruitment of A20, a complex deubiquitinating enzyme. Immunity 38: 896-905. doi: 10.1016/j.immuni.2013.03.008
|
| [99] |
Wertz IE, O'rourke KM, Zhou H, et al. (2004) De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-κB signalling. Nature 430: 694-699. doi: 10.1038/nature02794
|
| [100] |
Wertz IE, Newton K, Seshasayee D, et al. (2015) Phosphorylation and linear ubiquitin direct A20 inhibition of inflammation. Nature 528: 370-375. doi: 10.1038/nature16165
|
| [101] |
Mauro C, Pacifico F, Lavorgna A, et al. (2006) ABIN-1 binds to NEMO/IKKγ and co-operates with A20 in inhibiting NF-κB. J Biol Chem 281: 18482-18488. doi: 10.1074/jbc.M601502200
|
| [102] | Ha H, Han D, Choi Y (2009) TRAF-mediated TNFR-family signaling. Curr Protoc Immunol 87: 11. |
| [103] |
Heyninck K, Beyaert R (1999) The cytokine-inducible zinc finger protein A20 inhibits IL-1-induced NF-κB activation at the level of TRAF6. FEBS Lett 442: 147-150. doi: 10.1016/S0014-5793(98)01645-7
|
| [104] |
De A, Dainichi T, Rathinam CV, et al. (2014) The deubiquitinase activity of A 20 is dispensable for NF-κ B signaling. EMBO Rep 15: 775-783. doi: 10.15252/embr.201338305
|
| [105] |
Boone DL, Turer EE, Lee EG, et al. (2004) The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol 5: 1052-1060. doi: 10.1038/ni1110
|
| [106] |
Harhaj EW, Dixit VM (2011) Deubiquitinases in the regulation of NF-κB signaling. Cell Res 21: 22-39. doi: 10.1038/cr.2010.166
|
| [107] | Lin FT, Lin VY, Lin VTG, et al. (2016) TRIP6 antagonizes the recruitment of A20 and CYLD to TRAF6 to promote the LPA2 receptor-mediated TRAF6 activation. Cell Discovery 2: 1-22. |
| [108] |
Saitoh T, Yamamoto M, Miyagishi M, et al. (2005) A20 is a negative regulator of IFN regulatory factor 3 signaling. J Immunol 174: 1507-1512. doi: 10.4049/jimmunol.174.3.1507
|
| [109] |
Feoktistova M, Makarov R, Brenji S, et al. (2020) A20 Promotes Ripoptosome Formation and TNF-Induced Apoptosis via cIAPs Regulation and NIK Stabilization in Keratinocytes. Cells 9: 351. doi: 10.3390/cells9020351
|
| [110] |
Li L, Huang B, Song S, et al. (2017) A20 functions as mediator in TNFα-induced injury of human umbilical vein endothelial cells through TAK1-dependent MAPK/eNOS pathway. Oncotarget 8: 65230. doi: 10.18632/oncotarget.18191
|
| [111] |
Li Y, Mooney EC, Holden SE, et al. (2019) A20 orchestrates inflammatory response in the oral mucosa through restraining NF-κB activity. J Immunol 202: 2044-2056. doi: 10.4049/jimmunol.1801286
|
| [112] |
Li Y, Mooney EC, Xia XJ, et al. (2020) A20 restricts inflammatory response and desensitizes gingival keratinocytes to apoptosis. Front Immunol 11: 365. doi: 10.3389/fimmu.2020.00365
|
| [113] |
Martens A, Priem D, Hoste E, et al. (2020) Two distinct ubiquitin-binding motifs in A20 mediate its anti-inflammatory and cell-protective activities. Nat Immunol 21: 381-387. doi: 10.1038/s41590-020-0621-9
|
| [114] |
Soni D, Wang DM, Regmi SC, et al. (2018) Deubiquitinase function of A20 maintains and repairs endothelial barrier after lung vascular injury. Cell Death Discovery 4: 1-14. doi: 10.1038/s41420-018-0056-3
|
| [115] |
Mohebiany AN, Ramphal NS, Karram K, et al. (2020) Microglial A20 protects the brain from CD8 T-cell-mediated immunopathology. Cell Rep 30: 1585-1597. doi: 10.1016/j.celrep.2019.12.097
|
| [116] |
Ma A, Malynn BA (2012) A20: linking a complex regulator of ubiquitylation to immunity and human disease. Nat Rev Immunol 12: 774-785. doi: 10.1038/nri3313
|
| [117] |
Malynn BA, Ma A (2019) A20: a multifunctional tool for regulating immunity and preventing disease. Cell Immunol 340: 103914. doi: 10.1016/j.cellimm.2019.04.002
|
| [118] |
Hövelmeyer N, Reissig S, Thi Xuan N, et al. (2011) A20 deficiency in B cells enhances B-cell proliferation and results in the development of autoantibodies. Eur J Immunol 41: 595-601. doi: 10.1002/eji.201041313
|
| [119] |
Tavares RM, Turer EE, Liu CL, et al. (2010) The ubiquitin modifying enzyme A20 restricts B cell survival and prevents autoimmunity. Immunity 33: 181-191. doi: 10.1016/j.immuni.2010.07.017
|
| [120] |
Chu Y, Vahl JC, Kumar D, et al. (2011) B cells lacking the tumor suppressor TNFAIP3/A20 display impaired differentiation and hyperactivation and cause inflammation and autoimmunity in aged mice. Blood 117: 2227-2236. doi: 10.1182/blood-2010-09-306019
|
| [121] |
Hammer GE, Turer EE, Taylor KE, et al. (2011) Expression of A20 by dendritic cells preserves immune homeostasis and prevents colitis and spondyloarthritis. Nat Immunol 12: 1184-1193. doi: 10.1038/ni.2135
|
| [122] |
Matmati M, Jacques P, Maelfait J, et al. (2011) A20 (TNFAIP3) deficiency in myeloid cells triggers erosive polyarthritis resembling rheumatoid arthritis. Nat Genet 43: 908-912. doi: 10.1038/ng.874
|
| [123] |
Vereecke L, Sze M, Guire CM, et al. (2010) Enterocyte-specific A20 deficiency sensitizes to tumor necrosis factor-induced toxicity and experimental colitis. J Exp Med 207: 1513-1523. doi: 10.1084/jem.20092474
|
| [124] |
Mooney EC, Sahingur SE (2021) The ubiquitin system and A20: implications in health and disease. J Dent Res 100: 10-20. doi: 10.1177/0022034520949486
|
| [125] |
Razani B, Whang MI, Kim FS, et al. (2020) Non-catalytic ubiquitin binding by A20 prevents psoriatic arthritis–like disease and inflammation. Nat Immunol 21: 422-433. doi: 10.1038/s41590-020-0634-4
|
| [126] |
Yoon CI, Ahn SG, Bae SJ, et al. (2019) High A20 expression negatively impacts survival in patients with breast cancer. PLoS One 14: e0221721. doi: 10.1371/journal.pone.0221721
|
| [127] |
Lee YH, Song GG (2012) Associations between TNFAIP3 gene polymorphisms and systemic lupus erythematosus: a meta-analysis. Genet Test Mol Biomarkers 16: 1105-1110. doi: 10.1089/gtmb.2012.0096
|
| [128] |
Korman BD, Criswell LA (2015) Recent advances in the genetics of systemic sclerosis: toward biological and clinical significance. Curr Rheumatol Rep 17: 1-11. doi: 10.1007/s11926-014-0484-x
|
| [129] |
Mayes MD, Bossini-Castillo L, Gorlova O, et al. (2014) Immunochip analysis identifies multiple susceptibility loci for systemic sclerosis. Am J Hum Genet 94: 47-61. doi: 10.1016/j.ajhg.2013.12.002
|
| [130] |
Martin JE, Assassi S, Diaz-Gallo LM, et al. (2013) A systemic sclerosis and systemic lupus erythematosus pan-meta-GWAS reveals new shared susceptibility loci. Hum Mol Genet 22: 4021-4029. doi: 10.1093/hmg/ddt248
|
| [131] |
Dieude P, Guedj M, Wipff J, et al. (2010) Association of the TNFAIP3 rs5029939 variant with systemic sclerosis in the European Caucasian population. Ann Rheum Dis 69: 1958-1964. doi: 10.1136/ard.2009.127928
|
| [132] |
Koumakis E, Giraud M, Dieudé P, et al. (2013) SAT0013 Candidate gene study in systemic sclerosis identifies a rare and functional variant of TNFAIP3 locus as a risk factor for individual polyautoimmunity. Ann Rheum Dis 71: 475-475. doi: 10.1136/annrheumdis-2012-eular.1670
|
| [133] |
Liu Y, Ye Z, Li X, et al. (2017) Genetic and functional associations with decreased anti-inflammatory tumor necrosis factor alpha induced protein 3 in macrophages from subjects with axial spondyloarthritis. Front Immunol 8: 860. doi: 10.3389/fimmu.2017.00860
|
| [134] |
Sun YY, Fan YC, Wang N, et al. (2015) Increased A20 mRNA level in peripheral blood mononuclear cells is associated with immune phases of patients with chronic hepatitis B. Medicine 94: e2428. doi: 10.1097/MD.0000000000002428
|
| [135] |
Xu H, Wang L, Zheng P, et al. (2017) Elevated serum A20 is associated with severity of chronic hepatitis B and A20 inhibits NF-κB-mediated inflammatory response. Oncotarget 8: 38914. doi: 10.18632/oncotarget.17153
|
| [136] |
Hung YY, Lin CC, Kang HY, et al. (2017) TNFAIP3, a negative regulator of the TLR signaling pathway, is a potential predictive biomarker of response to antidepressant treatment in major depressive disorder. Brain Behav Immun 59: 265-272. doi: 10.1016/j.bbi.2016.09.014
|
| [137] |
Huang H, Tang QZ, Wang AB, et al. (2010) Tumor suppressor A20 protects against cardiac hypertrophy and fibrosis by blocking transforming growth factor-β-activated kinase 1-dependent signaling. Hypertension 56: 232-239. doi: 10.1161/HYPERTENSIONAHA.110.149963
|
| [138] |
Xu W, Wang C, Liang M, et al. (2018) A20 prevents obesity-induced development of cardiac dysfunction. J Mol Med 96: 159-172. doi: 10.1007/s00109-017-1608-3
|
| [139] | Jung SM, Lee JH, Park J, et al. (2013) Smad6 inhibits non-canonical TGF-β1 signalling by recruiting the deubiquitinase A20 to TRAF6. Nat Commun 4: 1-16. |
| [140] |
Liu S, Lv X, Liu C, et al. (2019) Targeting degradation of the transcription factor C/EBPβ reduces lung fibrosis by restoring activity of the ubiquitin-editing enzyme A20 in macrophages. Immunity 51: 522-534. doi: 10.1016/j.immuni.2019.06.014
|
| [141] |
Wang X, Ai L, Xu Q, et al. (2017) A20 attenuates liver fibrosis in NAFLD and inhibits inflammation responses. Inflammation 40: 840-848. doi: 10.1007/s10753-017-0528-2
|
| [142] | Kelly C, Reihill J, Malconsom B, et al. (2013) Defective A20 signalling in CF: Anti-inflammatory action of gibberellins. Eur Respir J 42: 2106. |
| [143] |
Malcomson B, Wilson H, Veglia E, et al. (2016) Connectivity mapping (ssCMap) to predict A20-inducing drugs and their antiinflammatory action in cystic fibrosis. P Natl Acad Sci USA 113: E3725-E3734. doi: 10.1073/pnas.1520289113
|
| [144] |
Bhattacharyya S, Wang W, Graham LVD, et al. (2016) A20 suppresses canonical Smad-dependent fibroblast activation: novel function for an endogenous inflammatory modulator. Arthritis Res Ther 18: 1-10. doi: 10.1186/s13075-016-1118-7
|
| [145] |
Hand LE, Usan P, Cooper GJS, et al. (2015) Adiponectin induces A20 expression in adipose tissue to confer metabolic benefit. Diabetes 64: 128-136. doi: 10.2337/db13-1835
|
| [146] |
Carrion AM, Link WA, Ledo F, et al. (1999) DREAM is a Ca2+-regulated transcriptional repressor. Nature 398: 80-84. doi: 10.1038/18044
|
| [147] |
Cebolla B, Fernández-Pérez A, Perea G, et al. (2008) DREAM mediates cAMP-dependent, Ca2+-induced stimulation of GFAP gene expression and regulates cortical astrogliogenesis. J Neurosci 28: 6703-6713. doi: 10.1523/JNEUROSCI.0215-08.2008
|
| [148] |
Savignac M, Mellström B, Bébin AG, et al. (2010) Increased B cell proliferation and reduced Ig production in DREAM transgenic mice. J Immunol 185: 7527-7536. doi: 10.4049/jimmunol.1000152
|
| [149] |
Tiruppathi C, Soni D, Wang DM, et al. (2014) The transcription factor DREAM represses the deubiquitinase A20 and mediates inflammation. Nat Immunol 15: 239-247. doi: 10.1038/ni.2823
|
| [150] |
Naranjo JR, Zhang H, Villar D, et al. (2016) Activating transcription factor 6 derepression mediates neuroprotection in Huntington disease. J Clin Invest 126: 627-638. doi: 10.1172/JCI82670
|
| [151] |
Lopez-Hurtado A, Peraza DA, Cercos P, et al. (2019) Targeting the neuronal calcium sensor DReAM with small-molecules for Huntington's disease treatment. Sci Rep 9: 1-16. doi: 10.1038/s41598-019-43677-7
|
| [152] |
Bhattacharyya S, Wang W, Tamaki Z, et al. (2018) Pharmacological inhibition of toll-like receptor-4 signaling by TAK242 prevents and induces regression of experimental organ fibrosis. Front Immunol 9: 2434. doi: 10.3389/fimmu.2018.02434
|
| [153] |
Bhattacharyya S, Wang W, Graham LVD, et al. (2016) A20 suppresses canonical Smad-dependent fibroblast activation: novel function for an endogenous inflammatory modulator. Arthritis Res Ther 18: 1-10. doi: 10.1186/s13075-016-1118-7
|


Figures(3) / Tables(1)

Swarna Bale, John Varga, Swati Bhattacharyya. Role of RP105 and A20 in negative regulation of toll-like receptor activity in fibrosis: potential targets for therapeutic intervention[J]. AIMS Allergy and Immunology, 2021, 5(2): 102-126. doi: 10.3934/Allergy.2021009







 DownLoad:
DownLoad: