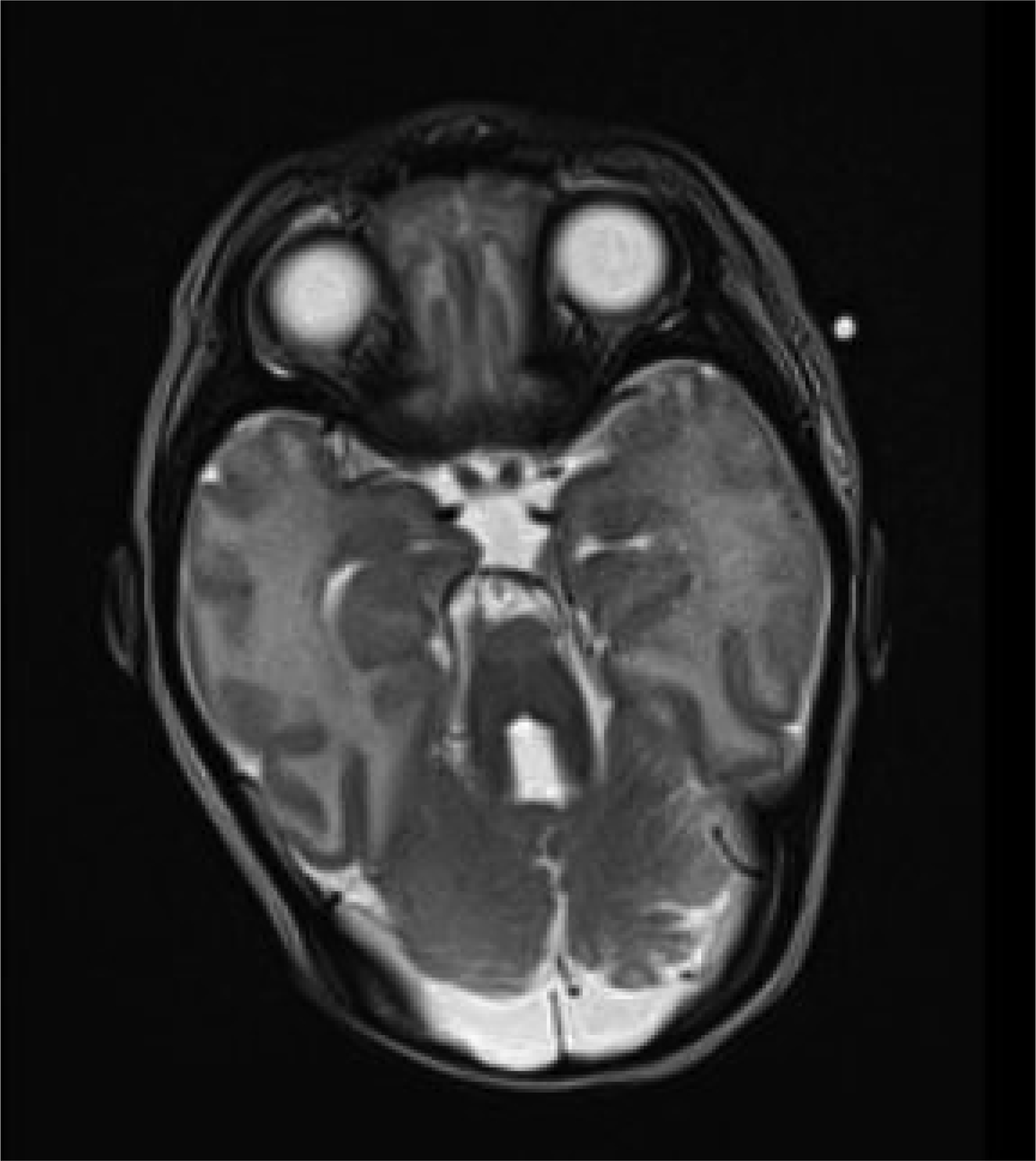
Joubert syndrome (JS) is a complex medical condition characterized by a pathognomonic midbrain-hindbrain malformation visible on brain imaging, which is known as the “molar tooth sign” (MTS). The presence of the MTS in the brain is the defining diagnostic criterion for JS. Individuals with JS commonly exhibit a developmental delay, hypotonia, and abnormal eye movements. In addition, neonatal breathing dysregulation is observed in about half of the cases. Midline brain defects associated with JS can lead to pituitary hormone abnormalities, thereby manifesting as multiple pituitary insufficiencies in the neonatal period, such as hypoglycemia and, in male patients, a micropenis with undescended testes. Although JS is a well-researched genetic condition, there is minimal information on the endocrinological aspects of JS. This manuscript aims to emphasize the spectrum of endocrinologic findings in JS through the retrospective evaluation of four cases characterized by combined pituitary dysfunctions, including secondary hypothyroidism, growth hormone deficiency, and panhypopituitarism. Highlighting these treatable aspects of JS is crucial, as continuous endocrinological monitoring can positively impact a patient's well-being, particularly in managing secondary adrenal and thyroid insufficiencies.
Citation: Elżbieta Marczak, Maria Szarras-Czapnik, Małgorzata Wójcik, Agata Zygmunt-Górska, Jerzy Starzyk, Karolina Czyżowska, Anna Szymańska, Katarzyna Gołąb-Jenerał, Agnieszka Zachurzok, Elżbieta Moszczyńska. Hypopituitarism—A rare manifestation in Joubert syndrome: about 4 cases[J]. AIMS Medical Science, 2024, 11(3): 318-329. doi: 10.3934/medsci.2024022
Joubert syndrome (JS) is a complex medical condition characterized by a pathognomonic midbrain-hindbrain malformation visible on brain imaging, which is known as the “molar tooth sign” (MTS). The presence of the MTS in the brain is the defining diagnostic criterion for JS. Individuals with JS commonly exhibit a developmental delay, hypotonia, and abnormal eye movements. In addition, neonatal breathing dysregulation is observed in about half of the cases. Midline brain defects associated with JS can lead to pituitary hormone abnormalities, thereby manifesting as multiple pituitary insufficiencies in the neonatal period, such as hypoglycemia and, in male patients, a micropenis with undescended testes. Although JS is a well-researched genetic condition, there is minimal information on the endocrinological aspects of JS. This manuscript aims to emphasize the spectrum of endocrinologic findings in JS through the retrospective evaluation of four cases characterized by combined pituitary dysfunctions, including secondary hypothyroidism, growth hormone deficiency, and panhypopituitarism. Highlighting these treatable aspects of JS is crucial, as continuous endocrinological monitoring can positively impact a patient's well-being, particularly in managing secondary adrenal and thyroid insufficiencies.
Adrenocorticotropic hormone
Central adrenal insufficiency
Continuous positive airway pressure
Dehydroepiandrosterone-sulfate
Follicle stimulating hormone
Free triiodothyronine
Free thyroxine
Gas chromatography–mass spectrometry
Growth hormone deficiency
Insulin-like growth factor 1
Insulin-like growth factor binding protein 3
Intramuscular injection
Joubert syndrome
Luteinizing hormone
Magnetic resonance imaging
Mass spectrometry
Molar tooth sign
Not applicable
Pituitary stalk interruption syndrome
Recombinant human growth hormone
Testosterone enanthate
Thyroid stimulating hormone
Thyroid hormones

| [1] |
Moore ER (2022) Primary cilia: the new face of craniofacial research. Biomolecules 12: 1724. https://doi.org/10.3390/biom12121724 
|
| [2] |
Stephen J, Vilboux T, Mian L, et al. (2017) Mutations in KIAA0753 cause Joubert syndrome associated with growth hormone deficiency. Hum Genet 136: 399-408. https://doi.org/10.1007/s00439-017-1765-z
|
| [3] |
Niceta M, Dentici ML, Ciolfi A, et al. (2020) Co-occurrence of mutations in KIF7 and KIAA0556 in Joubert syndrome with ocular coloboma, pituitary malformation and growth hormone deficiency: a case report and literature review. BMC Pediatr 20: 120. https://doi.org/10.1186/s12887-020-2019-0
|
| [4] |
Akcan N, Bas F, Poyrazoglu S, et al. (2019) Joubert syndrome with multiple pituitary hormone deficiency. BMJ Case Rep 12: e229016. https://doi.org/10.1136/bcr-2018-229016
|
| [5] |
Sanders AAWM, de Vrieze E, Alazami AM, et al. (2015) KIAA0556 is a novel ciliary basal body component mutated in Joubert syndrome. Genome Biol 16: 293. https://doi.org/10.1186/s13059-015-0858-z
|
| [6] |
Wolf MTF, Saunier S, O'Toole JF, et al. (2007) Mutational analysis of the RPGRIP1L gene in patients with Joubert syndrome and nephronophthisis. Kidney Int 72: 1520-1526. https://doi.org/10.1038/sj.ki.5002630
|
| [7] |
Marczak E, Szarras-Czapnik M, Moszczyńska E (2023) Endocrine manifestations in Joubert syndrome—literature review. AIMS Med Sci 10: 343-352. https://doi.org/10.3934/medsci.2023027
|
| [8] |
Delous M, Baala L, Salomon R, et al. (2007) The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat Genet 39: 875-881. https://doi.org/10.1038/ng2039
|
| [9] |
Bachmann-Gagescu R, Dempsey JC, Bulgheroni S, et al. (2020) Healthcare recommendations for Joubert syndrome. Am J Med Genet A 182: 229-249. https://doi.org/10.1002/ajmg.a.61399
|
| [10] |
Parisi MA, Doherty D, Chance PF, et al. (2007) Joubert syndrome (and related disorders) (OMIM 213300). Eur J Hum Genet 15: 511-521. https://doi.org/10.1038/sj.ejhg.5201648
|

Figures(2) / Tables(2)

Elżbieta Marczak, Maria Szarras-Czapnik, Małgorzata Wójcik, Agata Zygmunt-Górska, Jerzy Starzyk, Karolina Czyżowska, Anna Szymańska, Katarzyna Gołąb-Jenerał, Agnieszka Zachurzok, Elżbieta Moszczyńska. Hypopituitarism—A rare manifestation in Joubert syndrome: about 4 cases[J]. AIMS Medical Science, 2024, 11(3): 318-329. doi: 10.3934/medsci.2024022







 DownLoad:
DownLoad: