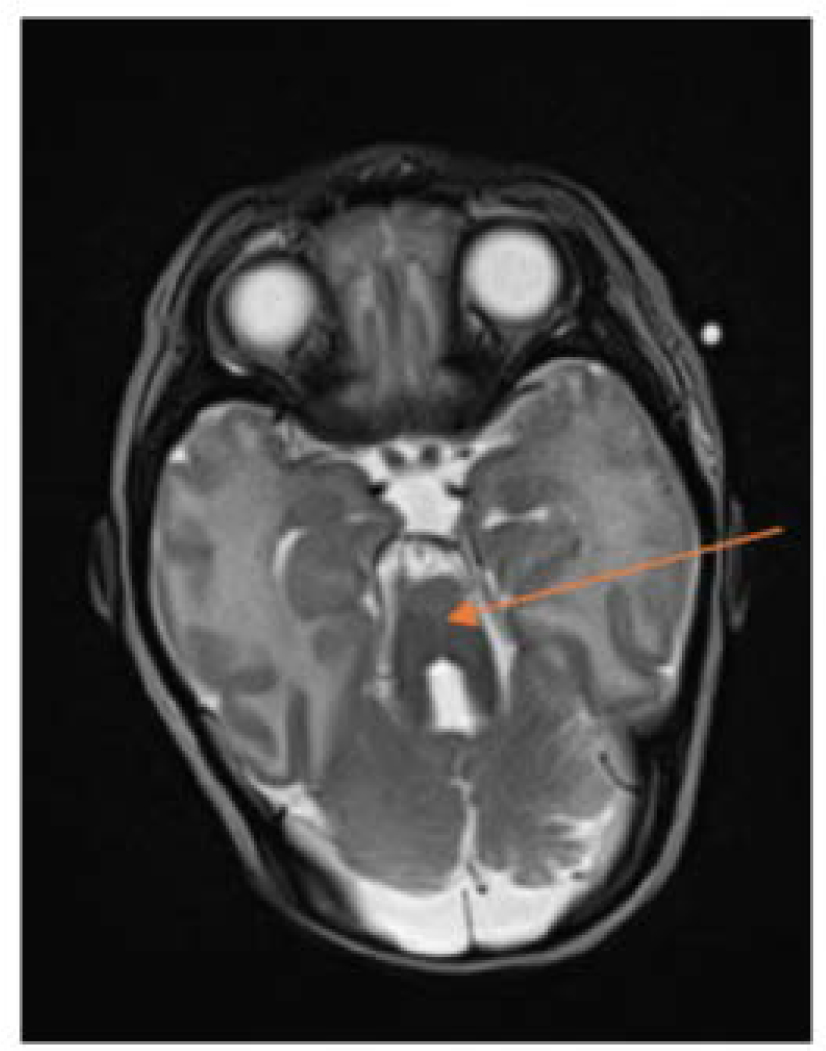
Joubert syndrome (JS) is a heterogeneously inherited, rare, autosomal recessive disorder characterised by neonatal breathing dysregulation, developmental delay, hypotonia, abnormal eye movements and a distinctive cerebellar and brainstem malformation called the molar tooth sign (MTS). Patients with JS may develop hypothalamic-pituitary dysfunction, leading to growth hormone deficiency, hypothyroidism, adrenal insufficiency and hypogonadism. This review summarizes the screening, diagnosis, and management of these conditions in JS.
Citation: Elzbieta Marczak, Maria Szarras-Czapnik, Elzbieta Moszczyńska. Endocrine manifestations in Joubert syndrome—literature review[J]. AIMS Medical Science, 2023, 10(4): 343-352. doi: 10.3934/medsci.2023027
Joubert syndrome (JS) is a heterogeneously inherited, rare, autosomal recessive disorder characterised by neonatal breathing dysregulation, developmental delay, hypotonia, abnormal eye movements and a distinctive cerebellar and brainstem malformation called the molar tooth sign (MTS). Patients with JS may develop hypothalamic-pituitary dysfunction, leading to growth hormone deficiency, hypothyroidism, adrenal insufficiency and hypogonadism. This review summarizes the screening, diagnosis, and management of these conditions in JS.

| [1] |
Brancati F, Dallapiccola B, Valente EM (2010) Joubert syndrome and related disorders. Orphanet J Rare Dis 5: 20. https://doi.org/10.1186/1750-1172-5-20 
|
| [2] |
Guemez-Gamboa A, Coufal NG, Gleeson JG (2014) Primary cilia in the developing and mature brain. Neuron 82: 511-521. https://doi.org/10.1016/j.neuron.2014.04.024
|
| [3] |
Parisi MA (2009) Clinical and molecular features of Joubert syndrome and related disorders. Am J Med Genet C Semin Med Genet 151: 326-340. https://doi.org/10.1002/ajmg.c.30229
|
| [4] |
Bachmann-Gagescu R, Dempsey JC, Bulgheroni S, et al. (2020) Healthcare recommendations for Joubert syndrome. Am J Med Genet A 182: 229-249. https://doi.org/10.1002/ajmg.a.61399
|
| [5] | Parisi M, Glass I (2003) Joubert syndromesyndrome, In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet] . Seattle: University of Washington. |
| [6] |
Poretti A, Boltshauser E, Loenneker T, et al. (2007) Diffusion tensor imaging in Joubert syndrome. Am J Neuroradiol 28: 1929-1933. https://doi.org/10.3174/ajnr.A0703
|
| [7] |
Wolf MTF, Saunier S, O'Toole JF, et al. (2007) Mutational analysis of the RPGRIP1L gene in patients with Joubert syndrome and nephronophthisis. Kidney Int 72: 1520-1526. https://doi.org/10.1038/sj.ki.5002630
|
| [8] |
Aljeaid D, Lombardo RC, Witte DP, et al. (2019) A novel pathogenic variant in OFD1 results in X-linked Joubert syndrome with orofaciodigital features and pituitary aplasia. Am J Med Genet A 179: 1010-1014. https://doi.org/10.1002/ajmg.a.61018
|
| [9] |
Sanders AAWM, de Vrieze E, Alazami AM, et al. (2015) KIAA0556 is a novel ciliary basal body component mutated in Joubert syndrome. Genome Biol 16: 293. https://doi.org/10.1186/s13059-015-0858-z
|
| [10] |
Gana S, Serpieri V, Valente EM (2022) Genotype–phenotype correlates in Joubert syndrome: A review. Am J Med Genet C Semin Med Genet 190: 72-88. https://doi.org/10.1002/ajmg.c.31963
|
| [11] |
Niceta M, Dentici ML, Ciolfi A, et al. (2020) Co-occurrence of mutations in KIF7 and KIAA0556 in Joubert syndrome with ocular coloboma, pituitary malformation and growth hormone deficiency: A case report and literature review. BMC Pediatr 20: 120. https://doi.org/10.1186/s12887-020-2019-0
|
| [12] |
Lee JE, Silhavy JL, Zaki MS, et al. (2012) CEP41 is mutated in Joubert syndrome and is required for tubulin glutamylation at the cilium. Nat Genet 44: 193-199. https://doi.org/10.1038/ng.1078
|
| [13] |
Van De Weghe JC, Rusterholz TDS, Latour B, et al. (2017) Mutations in ARMC9, which encodes a basal body protein, cause Joubert syndrome in humans and ciliopathy phenotypes in Zebrafish. Am J Hum Genet 101: 23-36. https://doi.org/10.1016/j.ajhg.2017.05.010
|
| [14] |
Delous M, Baala L, Salomon R, et al. (2007) The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat Genet 39: 875-881. https://doi.org/10.1038/ng2039
|
| [15] |
Stephen J, Vilboux T, Mian L, et al. (2017) Mutations in KIAA0753 cause Joubert syndrome associated with growth hormone deficiency. Hum Genet 136: 399-408. https://doi.org/10.1007/s00439-017-1765-z
|
| [16] |
Akcan N, Bas F, Poyrazoglu S, et al. (2019) Joubert syndrome with multiple pituitary hormone deficiency. BMJ Case Rep 12: e229016. https://doi.org/10.1136/bcr-2018-229016
|
| [17] |
Vilboux T, Malicdan MCV, Roney JC, et al. (2017) CELSR2, encoding a planar cell polarity protein, is a putative gene in Joubert syndrome with cortical heterotopia, microophthalmia, and growth hormone deficiency. Am J Med Genet A 173: 661-666. https://doi.org/10.1002/ajmg.a.38005
|
| [18] |
Parisi MA, Doherty D, Chance PF, et al. (2007) Joubert syndrome (and related disorders) (OMIM 213300). Eur J Hum Genet 15: 511-521. https://doi.org/10.1038/sj.ejhg.5201648
|
| [19] |
Lucas-Herald AK, Kinning E, Iida A, et al. (2015) A case of functional growth hormone deficiency and early growth retardation in a child with IFT172 mutations. J Clin Endocrinol Metab 100: 1221-1224. https://doi.org/10.1210/jc.2014-3852
|
| [20] |
Hardee I, Soldatos A, Davids M, et al. (2017) Defective ciliogenesis in INPP5E-related Joubert syndrome. Am J Med Genet A 173: 3231-3237. https://doi.org/10.1002/ajmg.a.38376
|
| [21] | Parisi MA, Pinter JD, Glass IA, et al. (2004) Cerebral and cerebellar motor activation abnormalities in a subject with Joubert syndrome: functional magnetic resonance imaging (MRI) study. J Child Neurol 19: 214-218. |
| [22] |
Bachmann-Gagescu R, Dempsey JC, Phelps IG, et al. (2015) Joubert syndrome: A model for untangling recessive disorders with extreme genetic heterogeneity. J Med Genet 52: 514-522. https://doi.org/10.1136/jmedgenet-2015-103087
|
| [23] | Hawkes CP, Grimberg A (2013) Measuring growth hormone and insulin-like growth factor-I in infants: What is normal?. Pediatr Endocrinol Rev 11: 126-146. |
| [24] |
Gujral J, Yau M, Yang AC, et al. (2019) Primary cortisol deficiency and growth hormone deficiency in a neonate with hypoglycemia: coincidence or consequence?. J Endocr Soc 3: 838-846. https://doi.org/10.1210/js.2018-00386
|
| [25] | Rosenfeld E, Thornton PS (2023) Hypoglycemia in Neonates Infants, and Children, In: Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet] . South Dartmouth: MDText.com, Inc. |
| [26] |
Dempsher D (2008) Adrenal and pituitary insufficiency in the neonate. NeoReviews 9. https://doi.org/10.1542/neo.9-2-e72
|
| [27] |
Yiğit Ş, Türkmen M, Tuncer O, et al. (2018) Neonatal adrenal insufficiency: Turkish neonatal and pediatric endocrinology and diabetes societies consensus report. Turk Pediatri Ars 53: S239-S243. https://doi.org/10.5152/TurkPediatriArs.2018.01822
|
| [28] |
Vigneri R, D'Agata R (1971) Growth hormone release during the first year of life in relation to sleep-wake periods. J Clin Endocrinol Metab 33: 561-563. https://doi.org/10.1210/jcem-33-3-561
|
| [29] |
Hammarsjö A, Wang Z, Vaz R, et al. (2017) Novel KIAA0753 mutations extend the phenotype of skeletal ciliopathies. Sci Rep 7: 15585. https://doi.org/10.1038/s41598-017-15442-1
|
| [30] |
Jacoby M, Cox JJ, Gayral S, et al. (2009) INPP5E mutations cause primary cilium signaling defects, ciliary instability and ciliopathies in human and mouse. Nat Genet 41: 1027-1031. https://doi.org/10.1038/ng.427
|
| [31] |
Hampshire DJ, Ayub M, Springell K, et al. (2006) MORM syndrome (mental retardation, truncal obesity, retinal dystrophy and micropenis), a new autosomal recessive disorder, links to 9q34. Eur J Hum Genet 14: 543-548. https://doi.org/10.1038/sj.ejhg.5201577
|
| [32] |
Bielas SL, Silhavy JL, Brancati F, et al. (2009) Mutations in INPP5E, encoding inositol polyphosphate-5-phosphatase E, link phosphatidyl inositol signaling to the ciliopathies. Nat Genet 41: 1032-1036. https://doi.org/10.1038/ng.423
|
| [33] |
Roemmich JN, Huerta MG, Sundaresan SM, et al. (2001) Alterations in body composition and fat distribution in growth hormone-deficient prepubertal children during growth hormone therapy. Metabolism 50: 537-547. https://doi.org/10.1053/meta.2001.22510
|
| [34] |
Aslan H, Ulker V, Gulcan EM, et al. (2002) Prenatal diagnosis of Joubert syndrome: A case report. Prenat Diagn 22: 13-16. https://doi.org/10.1002/pd.220
|
| [35] |
Uzunhan TA, Ertürk B, Aydın K, et al. (2023) Clinical and genetic spectrum from a prototype of ciliopathy: Joubert syndrome. Clin Neurol Neurosurg 224: 107560. https://doi.org/10.1016/j.clineuro.2022.107560
|
| [36] |
Sumathipala D, Strømme P, Gilissen C, et al. (2020) Sudden death in epilepsy and ectopic neurohypophysis in Joubert syndrome 23 diagnosed using SNVs/indels and structural variants pipelines on WGS data: A case report. BMC Med Genet 21: 96. https://doi.org/10.1186/s12881-020-01024-y
|
| [37] |
Graber JJ, Lau H, Sathe S (2009) Teaching neuroimages: molar tooth sign with hypotonia, ataxia, and nystagmus (Joubert Syndrome) and hypothyroidism. Neurology 73: e106. https://doi.org/10.1212/WNL.0b013e3181c679ba
|
Figures(1) / Tables(1)

Elzbieta Marczak, Maria Szarras-Czapnik, Elzbieta Moszczyńska. Endocrine manifestations in Joubert syndrome—literature review[J]. AIMS Medical Science, 2023, 10(4): 343-352. doi: 10.3934/medsci.2023027







 DownLoad:
DownLoad: