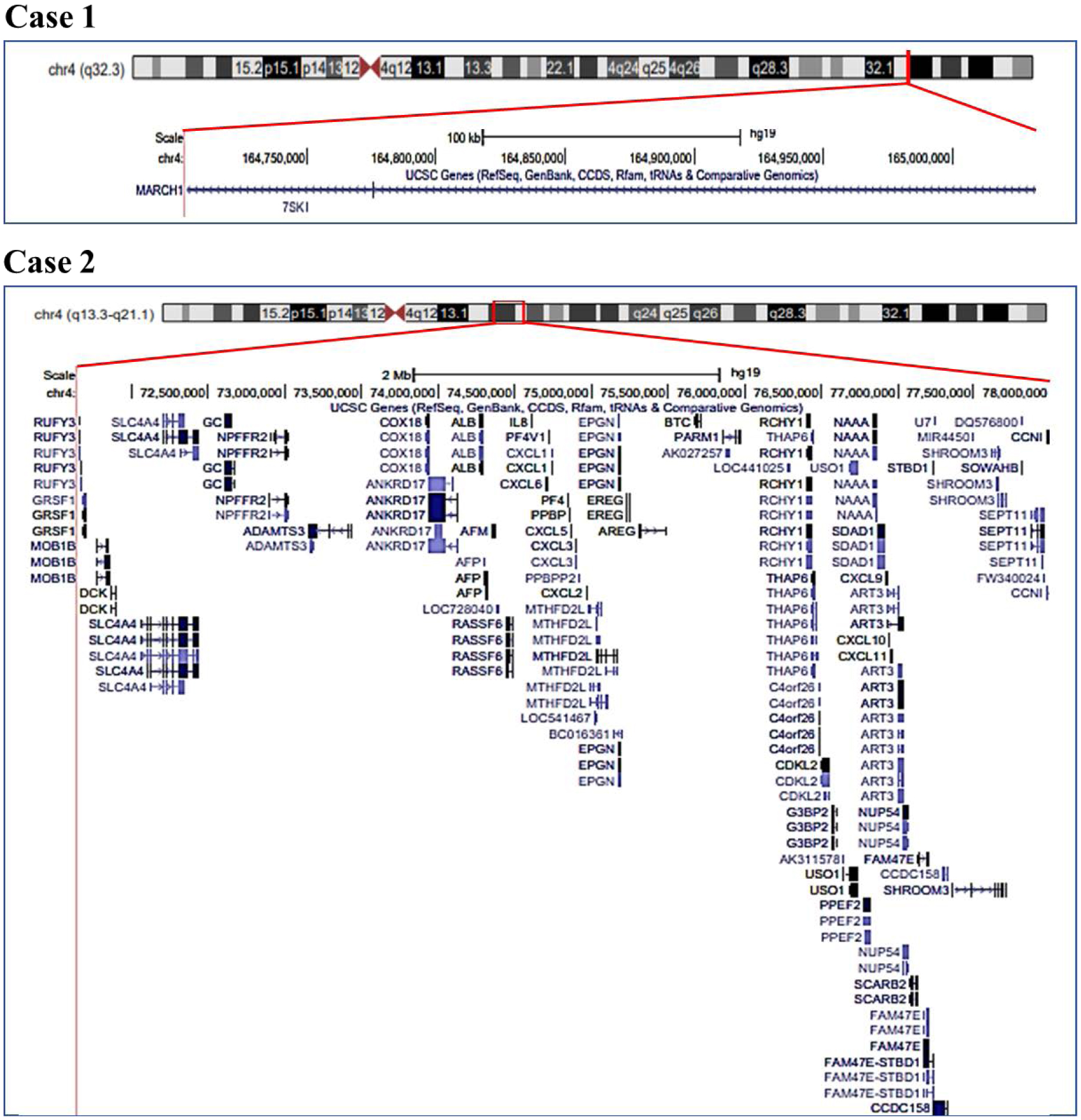
The 4q deletion syndrome defines a disorder, which may involve patients affected by either the deletion of the interstitial region from the centromere to 4q31 or by the deletion of the terminal region from 4q31 to 4qter. Here, we describe clinical phenotypes of two unrelated children of the same age followed at the same time, with case 1 presenting with 4q interstitial and case 2 with terminal 4q deletion, and compare them each other and with those reported in the literature. Both children showed complex, heterogeneous clinical manifestations, including craniofacial features, pre-postnatal growth failure, speech and developmental delay. In case 2, thyroid and cholesterol dysfunction were also found. Analyzing these data, clinical differences between interstitial and terminal 4q deletions are scanty and no significant phenotype differences were found between the 4q regions deleted as observed in the comparison of the two children and the related cases of the literature. The term 4q deletion syndrome - inclusive for both the interstitial and terminal 4q regions deleted - seems to be appropriate. To note, the dysfunction of cholesterol metabolism and thyroid presented by case 2 may be clinically worthwhile, whether confirmed by other observations.
Citation: Piero Pavone, Xena Giada Pappalardo, Riccardo Lubrano, Salvatore Savasta, Alberto Verrotti, Pasquale Parisi, Raffaele Falsaperla. 4q interstitial and terminal deletion: clinical features comparison in two unrelated children[J]. AIMS Medical Science, 2023, 10(2): 130-140. doi: 10.3934/medsci.2023011
The 4q deletion syndrome defines a disorder, which may involve patients affected by either the deletion of the interstitial region from the centromere to 4q31 or by the deletion of the terminal region from 4q31 to 4qter. Here, we describe clinical phenotypes of two unrelated children of the same age followed at the same time, with case 1 presenting with 4q interstitial and case 2 with terminal 4q deletion, and compare them each other and with those reported in the literature. Both children showed complex, heterogeneous clinical manifestations, including craniofacial features, pre-postnatal growth failure, speech and developmental delay. In case 2, thyroid and cholesterol dysfunction were also found. Analyzing these data, clinical differences between interstitial and terminal 4q deletions are scanty and no significant phenotype differences were found between the 4q regions deleted as observed in the comparison of the two children and the related cases of the literature. The term 4q deletion syndrome - inclusive for both the interstitial and terminal 4q regions deleted - seems to be appropriate. To note, the dysfunction of cholesterol metabolism and thyroid presented by case 2 may be clinically worthwhile, whether confirmed by other observations.

| [1] | Ockey CH, Feldman GV, Macaulay ME, et al. (1967) A large deletion of the long arm of chromosome No. 4 in a child with limb abnormalities. Arch Dis Child 42: 428-434. https://doi.org/10.1136/adc.42.224.428 |
| [2] | Golbus MS, Conte FA, Daentl DL (1973) Deletion from the long arm of chromosome 4 (46,XX,4q-) associated with congenital anomalies. J Med Genet 10: 83-85. https://doi.org/10.1136/jmg.10.1.83 |
| [3] | Townes PL, White M, Di Marzo SV (1979) 4q-syndrome. Am J Dis Child 133: 383-385. https://doi.org/10.1001/archpedi.1979.02130040037008 |
| [4] | Lin AE, Garver KL, Diggans G, et al. (1988) Interstitial and terminal deletions of the long arm of chromosome 4: further delineation of phenotypes. Am J Med Genet 31: 533-548. https://doi.org/10.1002/ajmg.1320310308 |
| [5] | Strehle EM, Ahmed OA, Hameed M, et al. (2001) The 4q-syndrome. Genet Couns 12: 327-339. |
| [6] | Strehle EM, Bantock HM (2003) The phenotype of patients with 4q-syndrome. Genet Couns 14: 195-205. |
| [7] | Strehle EM, Yu L, Rosenfeld JA, et al. (2012) Genotype-phenotype analysis of 4q deletion syndrome: proposal of a critical region. Am J Med Genet A 158A: 2139-2151. https://doi.org/10.1002/ajmg.a.35502 |
| [8] | Strehle EM, Gruszfeld D, Schenk D, et al. (2012) The spectrum of 4q- syndrome illustrated by a case series. Gene 506: 387-391. https://doi.org/10.1016/j.gene.2012.06.087 |
| [9] | Xu W, Ahmad A, Dagenais S, et al. (2012) Chromosome 4q deletion syndrome: narrowing the cardiovascular critical region to 4q32.2–q34.3. Am J Med Genet A 158A: 635-640. https://doi.org/10.1002/ajmg.a.34425 |
| [10] | Pappalardo XG, Ruggieri M, Falsaperla R, et al. (2021) A novel 4q32.3 deletion in a child: additional signs and the role of MARCH1. J Pediatr Genet 10: 259-265. https://doi.org/1055/s-0041-1736458 |
| [11] | Kuldeep CM, Khare AK, Garg A, et al. (2012) Terminal 4q deletion syndrome. Indian J Dermatol 57: 222-224. https://doi.org/10.4103/0019-5154.96203 |
| [12] | Sandal G, Ormeci AR, Oztas S (2013) De novo terminal 4q deletion syndrome with new ocular findings in Turkish twins: case report. Genet Couns 24: 217-222. |
| [13] | Vona B, Nanda I, Neuner C, et al. (2014) Terminal chromosome 4q deletion syndrome in an infant with hearing impairment and moderate syndromic features: review of literature. BMC Med Genet 15: 72. https://doi.org/10.1186/1471-2350-15-72 |
| [14] | Meaux T, Zeringue A, Mumphrey C, et al. (2015) Bilateral absence of the ulna in 4q terminal deletion syndrome: evidence for a critical region. Clin Dysmorphol 24: 122-124. https://doi.org/10.1097/MCD.0000000000000078 |
| [15] | Lurie IW (2016) A critical region for ulnar defects in patients with 4q deletions may be narrowed. Clin Dysmorphol 25: 133. https://doi.org/10.1097/MCD.0000000000000131 |
| [16] | Quadrelli R, Strehle EM, Vaglio A, et al. (2007) A girl with del(4)(q33) and occipital encephalocele: clinical description and molecular genetic characterization of a rare patient. Genet Test 11: 4-10. https://doi.org/10.1089/gte.2006.9995 |
| [17] | Zhang QS, Browning BL, Browning SR (2015) Genome-wide haplotypic testing in a Finnish cohort identifies a novel association with low-density lipoprotein cholesterol. Eur J Hum Genet 23: 672-677. https://doi.org/10.1038/ejhg.2014.105 |
| [18] | Maldžienė Ž, Vaitėnienė EM, Aleksiūnienė B, et al. (2020) A case report of familial 4q13.3 microdeletion in three individuals with syndromic intellectual disability. BMC Med Genomics 13: 63. https://doi.org/10.1186/s12920-020-0711-4 |
Figures(1) / Tables(2)

Piero Pavone, Xena Giada Pappalardo, Riccardo Lubrano, Salvatore Savasta, Alberto Verrotti, Pasquale Parisi, Raffaele Falsaperla. 4q interstitial and terminal deletion: clinical features comparison in two unrelated children[J]. AIMS Medical Science, 2023, 10(2): 130-140. doi: 10.3934/medsci.2023011







 DownLoad:
DownLoad: