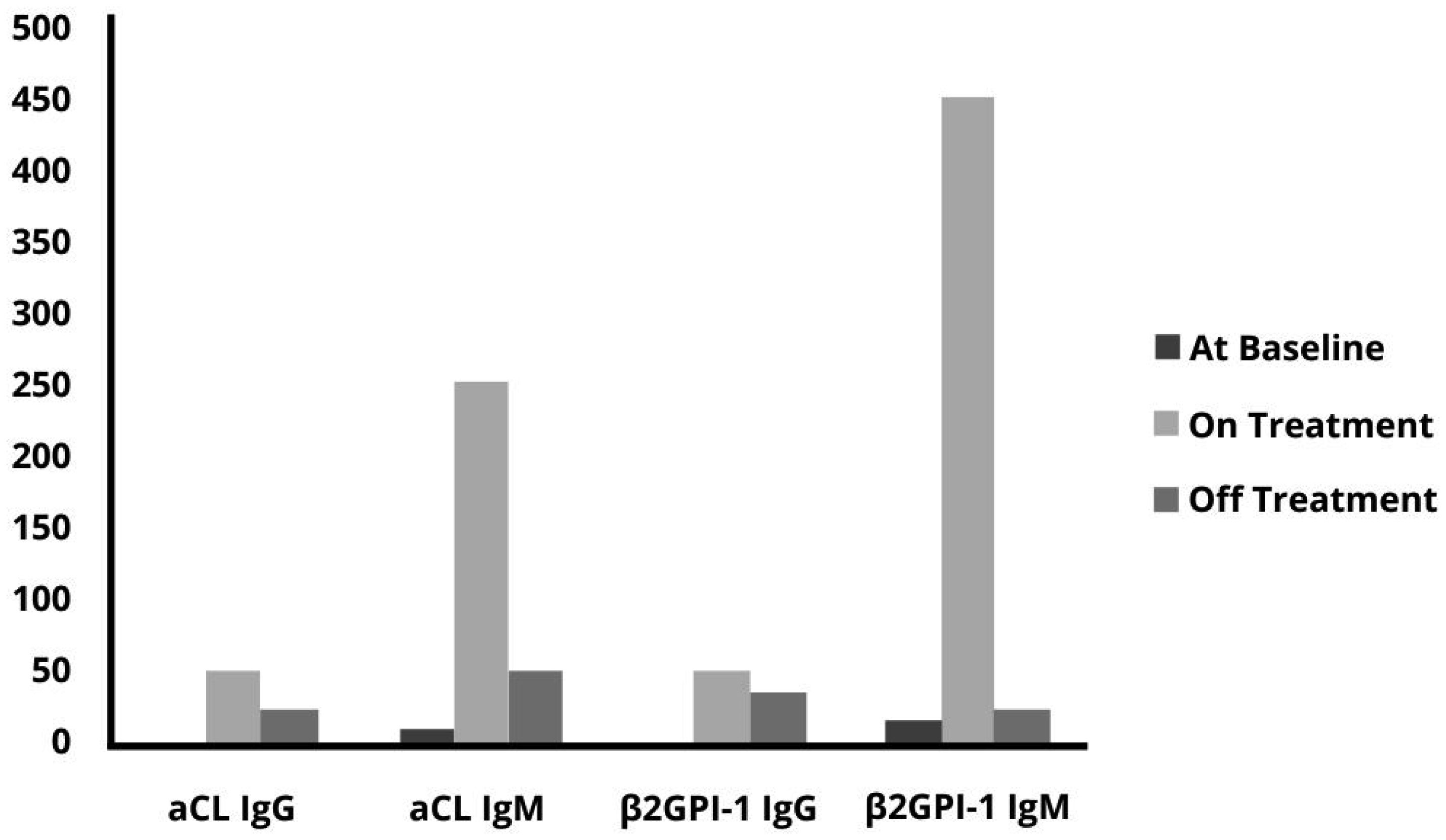
Sickle cell disease (SCD) is a common inherited condition in African, Caribbean, and Mediterranean countries. SCD is a hematologic disorder caused by a well-characterized point mutation in the β-globin gene, which produces an abnormal hemoglobin S that results in the sickling of red blood cells in deoxygenated conditions. Patients with SCD display a “hypercoagulation state” leading to an increased risk of severe venous and arterial thrombotic vascular events. Herein we report a case of severe thrombotic complications in a patient with SCD who showed high antiphospholipid antibodies (APA) levels during vascular occlusions.
Citation: Annamaria Petrungaro, Eugenia Quartarone, Luciana Rigoli, Paolo Sciarrone. Prothrombotic role of antiphospholipid antibodies in a patient with sickle cell disease[J]. AIMS Medical Science, 2022, 9(3): 362-366. doi: 10.3934/medsci.2022017
Sickle cell disease (SCD) is a common inherited condition in African, Caribbean, and Mediterranean countries. SCD is a hematologic disorder caused by a well-characterized point mutation in the β-globin gene, which produces an abnormal hemoglobin S that results in the sickling of red blood cells in deoxygenated conditions. Patients with SCD display a “hypercoagulation state” leading to an increased risk of severe venous and arterial thrombotic vascular events. Herein we report a case of severe thrombotic complications in a patient with SCD who showed high antiphospholipid antibodies (APA) levels during vascular occlusions.

| [1] |
Piel FB, Steinberg MH, Rees DC (2017) Sickle cell disease. N Engl J Med 376: 1561-1573. https://doi.org/10.1056/NEJMra1510865 
|
| [2] |
Noubiap JJ, Temgoua MN, Tankeu R, et al. (2018) Sickle cell disease, sickle trait and the risk for venous thromboembolism: a systematic review and meta-analysis. Thromb J 16: 27. https://doi.org/10.1186/s12959-018-0179-z
|
| [3] |
Conran N, De Paula EV (2020) Thromboinflammatory mechanisms in sickle cell disease—challenging the hemostatic balance. Haematologica 105: 2380-2390. https://doi.org/10.3324/haematol.2019.239343
|
| [4] |
Dowling MM, Quinn CT, Rogers ZR, et al. (2009) Stroke in sickle cell anemia: alternative etiologies. Pediatr Neurol 41: 124-126. https://doi:10.1016/j.pediatrneurol.2009.02.011
|
| [5] |
Aygun B, Padmanabhan S, Paley C, et al. (2002) Clinical significance of RBC alloantibodies and autoantibodies in sickle cell patients who received transfusions. Transfusion 42: 37-43. https://doi.org/10.1046/j.1537-2995.2002.00007.x
|
| [6] | Nsiri B, Ghazouani E, Gritli N, et al. (1998) Antiphospholipid antibodies: lupus anticoagulants, anticardiolipin and antiphospholipid isotypes in patients with sickle cell disease. Hematol Cell Ther 40: 107-112. |
| [7] |
Olayemi EE, Bazuaye GN (2009) Lupus anticoagulant and leg ulcers in sickle cell anemia. Indian J Dermatol 54: 251-254. https://doi.org/10.4103/0019-5154.55635
|
| [8] | Diatta A, Touré-Fall AO, Sarr NG, et al. (2004) Prevalence of antiphospholipid antibodies in patients with sickle cell disease. Ann Biol Clin (Paris) 62: 291-294. |
| [9] |
Hannemann A, Rees DC, Brewin JN, et al. (2018) Oxidative stress and phosphatidylserine exposure in red cells from patients with sickle cell anaemia. Br J Haematol 182: 567-578. https://doi.org/10.1111/bjh.15441
|
| [10] |
Merashli M, Arcaro A, Graf M, et al. (2021) Antiphospholipid antibodies in sickle cell disease: A systematic review and exploratory meta-analysis. Clin Appl Thromb Hemost 27: 10760296211002914. https://doi.org/10.1177/10760296211002914
|
| [11] |
Robazzi TC, Alves C, Abreu L, et al. (2015) Coexisting systemic lupus erythematosus and sickle cell disease: case report and literature review. Rev Bras Reumatol 55: 68-74. https://doi.org/10.1016/j.rbr.2013.05.005
|
| [12] | Christiaens C, Florkin B, Philippet P (2020) Coexistence of sickle cell disease and systemic lupus erythematosus. Rev Med Liege 75: 115-120. |
| [13] |
De Ceulaer K, Khamashta MA, Harris EN, et al. (1992) Antiphospholipid antibodies in homozygous sickle cell disease. Ann Rheum Dis 51: 671-672. https://doi.org/10.1136/ard.51.5.671
|
| [14] |
Kucuk O, Gilman-Sachs A, Beaman K, et al. (1993) Antiphospholipid antibodies in sickle cell disease. Am J Hematol 42: 380-383. https://doi.org/10.1002/ajh.2830420409
|
| [15] |
Piccin A, O'Connor-Byrne N, Daves M, et al. (2022) Autoimmune disease and sickle cell anaemia: “Intersecting pathways and differential diagnosis”. Br J Haematol 197: 518-528. https://doi.org/10.1111/bjh.18109
|
Figures(1)

Annamaria Petrungaro, Eugenia Quartarone, Luciana Rigoli, Paolo Sciarrone. Prothrombotic role of antiphospholipid antibodies in a patient with sickle cell disease[J]. AIMS Medical Science, 2022, 9(3): 362-366. doi: 10.3934/medsci.2022017







 DownLoad:
DownLoad: