Citation: Zhimin Wu, Tin Phan, Javier Baez, Yang Kuang, Eric J. Kostelich. Predictability and identifiability assessment of models for prostate cancerunder androgen suppression therapy[J]. Mathematical Biosciences and Engineering, 2019, 16(5): 3512-3536. doi: 10.3934/mbe.2019176

| [1] | C. Huggins and C. V. Hodges, Studies on Prostatic Cancer: I. The Effect of Castration, of |
| [2] | W. G. Nelson, Commentary on Huggins and Hodges: "Studies on Prostatic Cancer", Cancer Res., 76 (2016), 186–187. |
| [3] | S. Kumas, M. Shelley, C. Harrison, et al., Neo-adjuvant and adjuvant hormone therapy for localized and locally advanced prostate cancer, The Cochrane Database Syst. Rev., 18 (2006), CD006019. |
| [4] | B. J. Feldman and D. Feldman, The development of androgen-independent prostate cancer, Nat. Rev. Cancer, 1 (2001), 34–45. |
| [5] | R. Siegel, E. Ward, O. Brawley, et al., Cancer statistics, 2011: The impact of eliminating socioeconomic and racial disparities on premature cancer deaths, CA: A cancer journal for clinicians, 61 (2011) , 212–236. |
| [6] | N. A. Spry, L. Kristjanson, B. Hooton, et al., Adverse effects to quality of life arising from treatment can recover with intermittent androgen suppression in men with prostate cancer, Eur. J. Cancer, 42 (2006), 1083–1092. |
| [7] | N. D. Shore and E. D. Crawford, Intermittent Androgen Deprivation Therapy: Redefining the Standard of Care? Rev. Urol., 12 (2010), 1–11. |
| [8] | M. C. Eisenberg and H. V. Jain, A confidence building exercise in data and identifiability: Modeling cancer chemotherapy as a case study, J. Theor. Biol., 431 (2017), 63–78. |
| [9] | T. Phan, H. Changhan, A. Martinez, et al., Dynamics and implications of models for intermit- tent androgen suppression therapy, Math. Biosci. Eng., 16 (2019). |
| [10] | T. L. Jackson, A mathematical model of prostate tumor growth and androgen-independent re- lapse, Discrete Continuous Dyn. Syst. Ser. B, 4 (2003), 187–201. |
| [11] | A. M. Ideta, G. Tanaka, T. Takeuchi, et al., A mathematical model of intermittent androgen suppression for prostate cancer, J. Nonlinear Sci., 18 (2008), 593–614. |
| [12] | T. Portz, Y. Kuang and J. D. Nagy, A clinical data validated mathematical model of prostate cancer growth under intermittent androgen suppression therapy, AIP Adv., 2 (2012), 0–14. |
| [13] | H. Vardhan-Jain and A. Friedman, Modeling prostate cancer response to continuous versus intermittent androgen ablation therapy, Discrete & Continuous Dynamical Systems-Series B, 18 (2013) . |
| [14] | Y. Hirata, K. Akakura, C.S. Higano,et al., Quantitative mathematical modeling of PSA dy- namics of prostate cancer patients treated with intermittent androgen suppression, J. Mol. Cell. Biol., 4 (2012), 127–132. |
| [15] | Y. Hirata, N. Bruchovsky and K. Aihara, Development of a mathematical model that predicts the outcome of hormone therapy for prostate cancer, J. Theor. Biol., 264 (2010), 517–527. |
| [16] | Y. Hirata, G. Tanaka, N. Bruchovsky, et al., Mathematically modelling and controlling prostate cancer under intermittent hormone therapy, Asian J. Androl., 14 (2012), 270–277. |
| [17] | Q. Guo, Z. Lu, Y. Hirata, et al., Parameter estimation and optimal scheduling algorithm for a mathematical model of intermittent androgen suppression therapy for prostate cancer, Chaos, 23 (2013), 43125. |
| [18] | Y. Hirata, S. I. Azuma and K. Aihara, Model predictive control for optimally scheduling inter- mittent androgen suppression of prostate cancer, Methods, 67 (2014), 278–281. |
| [19] | Y. Hirata, K. Morino, K. Akakura, et al., Personalizing Androgen Suppression for Prostate Cancer Using Mathematical Modeling, Sci. Rep., 8 (2018), 2673. |
| [20] | R. A. Everett, A. M. Packer and Y. Kuang, Can Mathematical Models Predict the Outcomes of Prostate Cancer Patients Undergoing Intermittent Androgen Deprivation Therapy? Biophys. Rev. Lett., 9 (2014), 173–191. |
| [21] | M. Droop, Some thoughts on nutrient limitation in algae. J. Phycol., 9 (1973), 264–272. |
| [22] | J. Baez and Y. Kuang, Mathematical Models of Androgen Resistance in Prostate Cancer Pa- tients under Intermittent Androgen Suppression Therapy. Appl. Sci., 6 (2016), 352. |
| [23] | J. D. Morken, A. Packer, R. A. Everett, et al., Mechanisms of resistance to intermittent andro- gen deprivation in patients with prostate cancer identified by a novel computational method, Cancer Res., (2014). |
| [24] | T. Phan, K. Nguyen, P. Sharma, et al., The Impact of Intermittent Androgen Suppression Ther- apy in Prostate Cancer Modeling, Appl. Sci., 9 (2019), 36. |
| [25] | A. Zazoua and W. Wang, Analysis of mathematical model of prostate cancer with androgen deprivation therapy, Commun. Nonlinear Sci. Numer. Simul., 66 (2019), 41–60. |
| [26] | T. Hatano, Y. Hirata, H. Suzuki, et al., Comparison between mathematical models of intermit- tent androgen suppression for prostate cancer, J. Theor. Biol., 366 (2015), 33–45. |
| [27] | N. Bruchovsky, L. Klotz, J. Crook, et al., Final results of the Canadian prospective Phase II trial of intermittent androgen suppression for men in biochemical recurrence after radiotherapy for locally advanced prostate cancer: Clinical parameters, Cancer, 107 (2006), 389–395. |
| [28] | C. Cobelli and J. J .DiStefano-III, Parameter and structural identifiability concepts and ambigu- ities: a critical review and analysis, Am. J. Physiol. Regul. Integr. Comp. Physiol., 239 (1980), R7–24. |
| [29] | A. Raue, C. Kreutz, T. Maiwald, et al., Structural and practical identifiability analysis of partially observed dynamical models by exploiting the profile likelihood, Bioinformatics, 25 (2009), 1923–1929. |
| [30] | S. Audoly, G. Bellu, L. D'Angio, et al., Global identifiability of nonlinear models of biological systems, IEEE Trans. Biomed. Eng., 48 (2001), 55–65. |
| [31] | M. C. Eisenberg, S. L. Robertson and J. H. Tien, Identifiability and estimation of multiple transmission pathways in cholera and waterborne disease. J. Theor. Biol., 324 (2013), 84–102. |
| [32] | M. P. Saccomani and L. D'angiò, Examples of testing global identifiability with the DAISY software, IFAC Proc. Vol., 42 (2009), 48–53. |
| [33] | H. Wu, H. Zhu, H. Miao, et al., Parameter identifiability and estimation of HIV/AIDS dynamic models, Bull. Math. Biol., 70 (2008), 785–799. |
| [34] | M. C. Eisenberg and M. A. L. Hayashi, Determining identifiable parameter combinations using subset profiling, Math. Biosci., 256 (2014), 116–126. |
| [35] | H. Miao, X. Xia, A. S. Perelson, et al., On Identifiability of Nonlinear ODE Models and Ap- plications in Viral Dynamics, SIAM Rev. Soc. Ind. Appl. Math., 53 (2011), 3–39. |
| [36] | T. Quaiser and M. Monnigmann, Systematic identifiability testing for unambiguous mechanis- tic modeling - application to JAK-STAT, MAP kinase, and NF-κB signaling pathway models, BMC Syst. Biol., 3 (2009), 50. |
| [37] | C. Kreutz, A. Raue, D. Kaschek, et al., Profile likelihood in systems biology, FEBS J., 280 (2013), 2564–2571. |
| [38] | G. Evensen, Sequential data assimilation with a nonlinear quasi-geostrophic model using Monte Carlo methods to forecast error statistics, J. Geophys. Res. Oceans, 99 (1994), 10143– 10162. |
| [39] | G. Evensen, The ensemble Kalman filter: Theoretical formulation and practical implementa- tion, Ocean Dyn., 53 (2003), 343–367. |
| [40] | B. R. Hunt, E. J. Kostelich and I. Szunyogh, Efficient data assimilation for spatiotemporal chaos: A local ensemble transform Kalman filter, Physica D, 230 (2007), 112–126, |
| [41] | S. J. Baek, B. R. Hunt, E. Kalnay, et al., Local ensemble Kalman filtering in the presence of model bias, Tellus A, 58 (2006), 293–306. |
| [42] | C. P. Arnold and C. H. Day, Observing-Systems Simulation Experiments: Past, Present, and Future. Bull. Am. Meteorol. Soc., 67 (1986), 687–695. |
| [43] | R. M. Errico, R. Yang, N. C. Privé, et al., Development and validation of observing-system simulation experiments at NASA's Global Modeling and Assimilation Office, Q. J. R. Meteorol. Soc., 139 (2013), 1162–1178. |
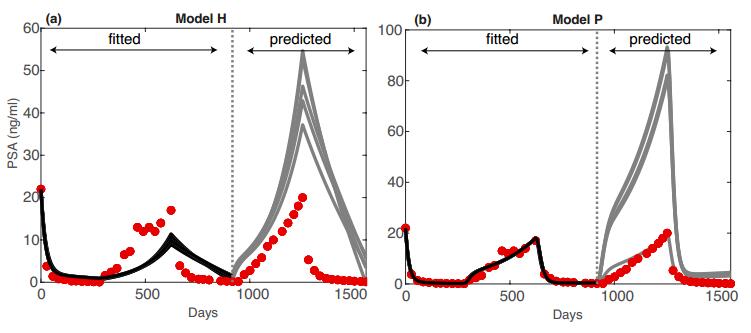
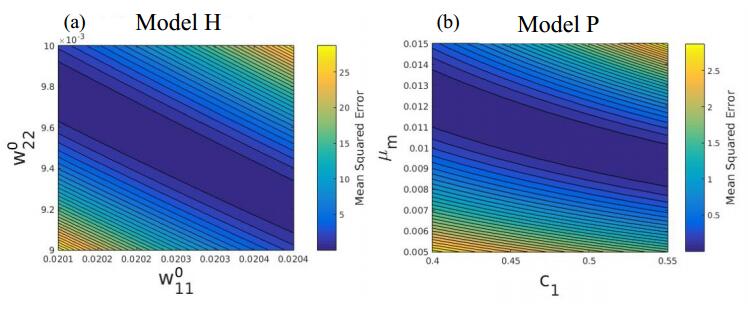

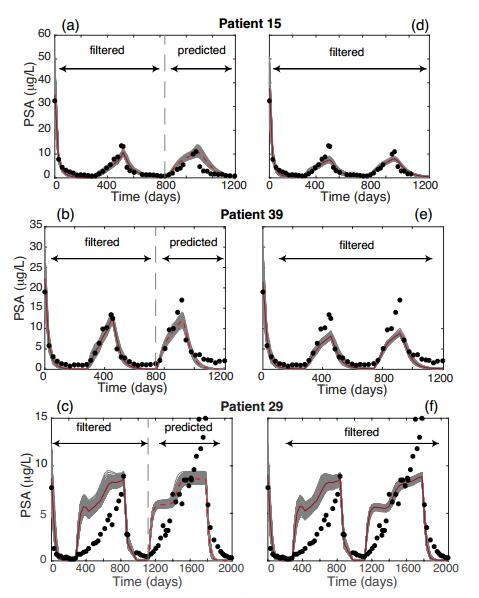
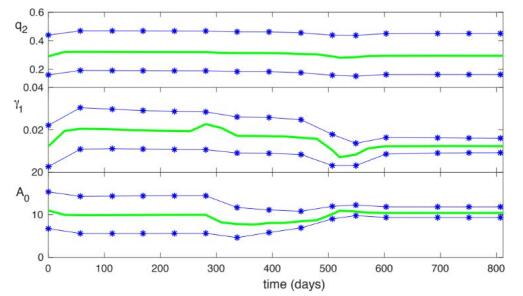
Figures(5) / Tables(4)

Zhimin Wu, Tin Phan, Javier Baez, Yang Kuang, Eric J. Kostelich. Predictability and identifiability assessment of models for prostate cancer under androgen suppression therapy[J]. Mathematical Biosciences and Engineering, 2019, 16(5): 3512-3536. doi: 10.3934/mbe.2019176







 DownLoad:
DownLoad: