Citation: Dong-Mei Li, Bing Chai, Yu-Li Fu, Qi Wang. Cytomegalovirus dynamics model with random behavior[J]. AIMS Mathematics, 2020, 5(6): 6373-6394. doi: 10.3934/math.2020410

| [1] | Q. X. Zeng, J. X. Dong, Y. Meng, et al. Progress in epidemiology of human cytomegalovirus Infection, Shandong medicine, 57 (2017), 1131-1133. |
| [2] | F. Chiuppesi, T. Kaltcheva, Z. Meng, et al., Identification of a continuous neutralizing epitope within UL128 of human cytomegalovirus, J. Virol., 91 (2017), 1-16. |
| [3] | S. E. Jackson, G. X. Sedikides, G. M. Mason, et al. Human Cytomegalovirus (HCMV)-Specific CD4+ T Cells are polyfunctional and can respond to HCMV-Infected dendritic cells in vitro, J. Virol., 91 (2017), 1-16. |
| [4] | D. Song, H. Mei, Research Progress of congenital cytomegalovirus infection in newborns, Medical Recapitulate, 23 (2017), 4453-4457. |
| [5] |
D. Zhu, C. Pan, J. Sheng, et al. Human cytomegalovirus reprogrammes haematopoietic progenitor cells into immunosuppressive monocytes to achieve latency, Nat. Microbiol., 3 (2018), 503-513. doi: 10.1038/s41564-018-0131-9

|
| [6] | Y. H. Li, Analysis of the correlation between breastfeeding of HCMV infected mothers and HCMV infection of newborns, MA. Thesis, Qingdao University, 2015. |
| [7] |
D. C. Moylan, S. K. Pati, S. A. Ross, et al. Breast milk HCMV viral load is associated with the establishment of breast milk CMV-pp65-specific CD8 T cells in Human CMV infected mothers, J. Infect. Dis., 216 (2017), 1176-1179. doi: 10.1093/infdis/jix457
|
| [8] | W. F. Wu, Progress in the treatment of CMV infection, Chinese Medical Journal, 38 (2003), 10-12. |
| [9] |
K. Wang, W. Wang, S. Song, Dynamics of an HBV model with diffusion and delay, J. Theor. Biol., 253 (2008), 36-44. doi: 10.1016/j.jtbi.2007.11.007
|
| [10] | G. Alter, D. Heckerman, A. Schneidewind, et al. HIV-1 adaptation to NK-cell-mediated immune pressure, Nature, 476 (2017), 96-100. |
| [11] | W. O. Kermack, A. G. Mckendrick, A contribution to the mathematical theory of epidemics, B. Math. Biol., 53 (1991), 57-87. |
| [12] | X. N. Han, The transmission dynamies of SARS, MA. Thesis, PLA Academy of Military Sciences, 2006. |
| [13] | R. Zhao, H. Wang, X. Wang, et al. Steroid therapy and the risk of osteonecrosis in SARS patients: a dose-response meta-analysis, Osteoporosis International, 28 (2016), 1027-1034. |
| [14] | L. F. Zhang, Comparison and parameter estimation between deterministic model and stochastic model of infectious disease transmission, MA. Thesis, Southwest Jiaotong University, 2010. |
| [15] | A. Q. Miao, J. Zhang, T. Zhang, et al. Threshold dynamics of a stochastic SIR model with vertical transmission and vaccination, Comput. Math. Method. M., 2017 (2017), 1-10. |
| [16] | P. Y. Xia, Dynamic behavior of several random virus models, Ph.D thesis, Northeast Normal University, 2018. |
| [17] | C. Y. Ji, Asymptotic behavior of stochastic biological model and infectious disease model, Ph.D thesis, Northeast Normal University, 2011. |
| [18] | Y. Asai, C. Tomás, X. Han, et al. A random model for immune response to virus in fluctuating environments, Springer International Publishing, 2016. |
| [19] | Y. Wang, J. Liu, Y. Y. Liu, et al. Establishment of mouse brain latent cytomegalovirus activation model, Progress in Modern Biomedicine, 15 (2015), 4414-4418. |
| [20] | H. Y. Duan, T. Yu, Diagnosis and treatment progress of cytomegalovirus infection, Chinese Journal of Obstetrics and Gynecology, 6 (2010), 68-71. |
| [21] |
M. A. Nowak, C. Bangham, Population dynamics of immune responses to persistent viruses, Science, 272 (1996), 74-79. doi: 10.1126/science.272.5258.74
|
| [22] |
M. A. Nowak, S. Bonhoeffer, A. M. Hill, et al. Viral dynamics in hepatitis B virus infection, Proceedings of the National Academy of Sciences, 93 (1996), 4398-4402. doi: 10.1073/pnas.93.9.4398
|
| [23] | X. Q. Niu, W. D. Li, G. F. Zhu, et al. Modeling the transmission dynamics of hepatitis B Virus and data assimilation forecasting, Mathematics in Practice and Theory, 45 (2015), 205-211. |
| [24] | J. M. Conway, D. Coombs, A stochastic model of latently infected cell reactivation and viral blip generation in treated HIV patients, PLoS Comput. Biol., 7 (2011), 1-24. |
| [25] |
C. Fraser, N. M. Ferguson, R. M. Anderson, et al. The role of antigenic stimulation and cytotoxic T Cell activity in regulating the long-term immunopathogenesis of HIV: mechanisms and clinical implications, Proceedings: Biological Sciences, 268 (2001), 2085-2095. doi: 10.1098/rspb.2001.1777
|
| [26] |
C. Fraser, N. M. Ferguson, R. M. Anderson, Quantification of intrinsic residual viral replication in treated HIV-infected patients, Proceedings of the National Academy of Sciences of the United States of America, 98 (2001), 15167-15172. doi: 10.1073/pnas.261283598
|
| [27] |
W. Zhang, L. M. Wahl, P. Yu, Viral blips may not need a trigger: how transient viremia can arise in deterministic in-host models, Siam Rev., 56 (2014), 127-155. doi: 10.1137/130937421
|
| [28] |
S. Wang, J. Zhang, F. Xu, et al. Dynamics of virus infection models with density‐dependent diffusion, Comput. Math. Appl., 74 (2017), 1-20. doi: 10.1016/j.camwa.2017.05.001
|
| [29] | M. Wei, L. Hu, X. Mao, Neutral stochastic functional differential equations with Lévy jumps under the local Lipschitz condition, Advances in Difference Equations, 2017 (1017), 57. |
| [30] | R. Khasminskii, Stochastic Stability of Differential Equations, Stochastic stability of differential equations, 1980. |
| [31] | X. X. Liao, Theory methods and application of sability, 2nd Edition, Huazhong University of Science and Technology Press, Wuhan, 2010. |
| [32] | N. He, W. D. Wang, A. R. Zhou, et al. Dynamics of stochastic HIV model based on saturation incidence rate, Journal of Southwest University (Natural Science Edition), 40 (2018), 123-125. |
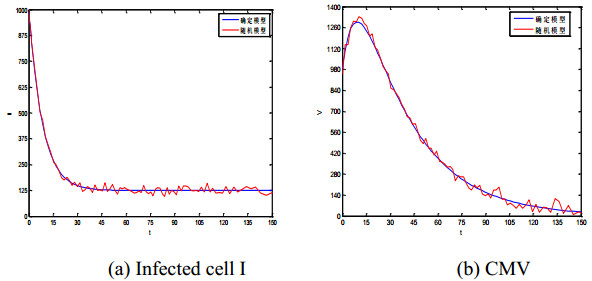
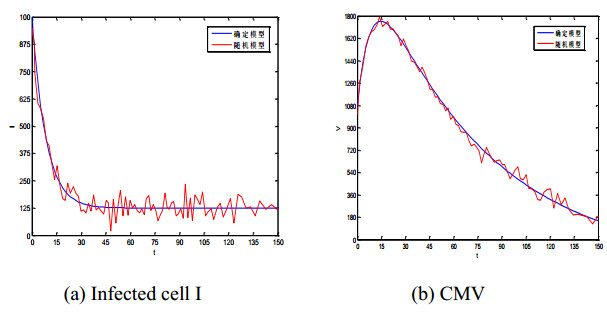
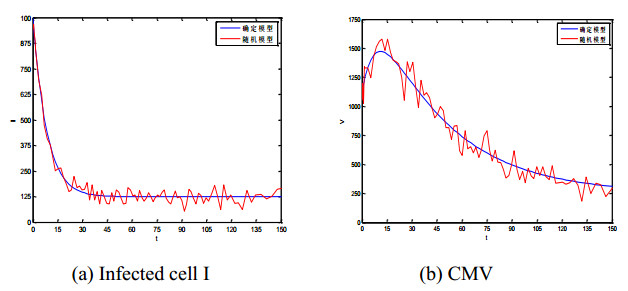
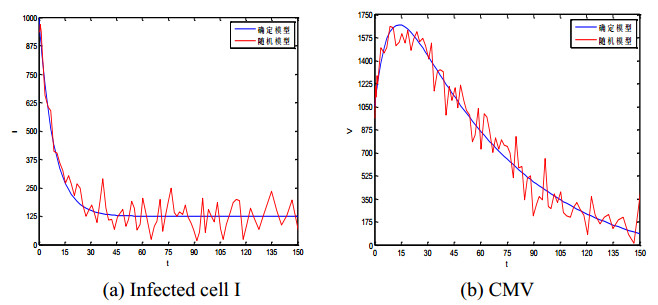
Figures(5) / Tables(1)

Dong-Mei Li, Bing Chai, Yu-Li Fu, Qi Wang. Cytomegalovirus dynamics model with random behavior[J]. AIMS Mathematics, 2020, 5(6): 6373-6394. doi: 10.3934/math.2020410







 DownLoad:
DownLoad: