Citation: Radi A. Jishi. Modified Becke-Johnson exchange potential: improved modeling of lead halides for solar cell applications[J]. AIMS Materials Science, 2016, 3(1): 149-159. doi: 10.3934/matersci.2016.1.149

| [1] |
Kojima A, Teshima K, Shirai Y, et al. (2009) Organometal halide perovskites as visible-light sensitizers for photovoltaic cells. J Am Chem Soc 131: 6050–6051. doi: 10.1021/ja809598r

|
| [2] |
Etgar L, Gau P, Xue Z, et al. (2012) Mesoscopic CH3NH3PbI3/TiO2 heterojunction solar cells. J Am Chem Soc 134: 17396–17399. doi: 10.1021/ja307789s
|
| [3] |
Ball J, Lee M, Hey A, et al. (2013) Low-temperature processed meso-superstructured to thin-film perovskite solar cells. Energy Env Sci 6: 1739–1743. doi: 10.1039/c3ee40810h
|
| [4] |
Heo H, Im S, Noh J, et al. (2013) Efficient inorganic-organic hybrid heterojunction solar cells containing perovskite compound and polymeric hole conductors. Nat Photonics 7: 486–491. doi: 10.1038/nphoton.2013.80
|
| [5] | Kim H-S, Lee J-W, Yantara N, et al. (2013) High efficiency solid-state sensitized solar cell based on submicrometer rutile TiO2 nanorod and CH3NH3PbI3 perovskite sensitizer. Nano Lett 13: 2412–2417. |
| [6] | Bi D, Yang L, Boschloo G, et al. (2013) Effect of different hole transport materials on recombination in CH3NH3PbI3 perovskite-sensitized mesoscopic solar cells. J Phys Chem Lett 4: 1532–1536. |
| [7] |
Cai B, Xing Y, Yang Z, et al. (2013) High performance hybrid solar cells sensitized by organolead halide perovskites. Energy Env Sci 6: 1480–1485. doi: 10.1039/c3ee40343b
|
| [8] |
Eperon G, Burlakov V, Docampo P, et al. (2014) Morphological control for high performance, solution-processed planar heterojunction perovskite solar cells. Adv Funct Mater 24: 151–157. doi: 10.1002/adfm.201302090
|
| [9] | Laban W, Etgar L. (2014) Depleted hole conductor-free lead halide iodide heterojunction solar cells. Energy Env Sci 6: 3249–3253. |
| [10] |
Stranks S, Eperon G, Grancini G, et al. (2013) Electron-hole diffusion lengths exceeding 1 micrometer in an organometal trihalide perovskite absorber. Science 342: 341–344. doi: 10.1126/science.1243982
|
| [11] |
Lee M, Teuscher J, Miyasaka T, et al. (2012) Efficient hybrid solar cells based on mesosuperstructured organometal halide perovskites. Science 338: 643–647. doi: 10.1126/science.1228604
|
| [12] | Noh J, Im S, Heo J, et al. (2013) Chemical management for colorful, efficient, and stable inorganicorganic hybrid nanostructured solar cells. Nano Lett 13: 1764–1769. |
| [13] |
Burschka J, Pellet N, Moon S, et al. (2013) Sequential deposition as a route to high-performance perovskite-sensitized solar cells. Nature 499: 316–319. doi: 10.1038/nature12340
|
| [14] |
Liu M, Johnston M, Snaith H (2013) Efficient planar heterojunction perovskite solar cells by vapour deposition. Nature 501: 395–398. doi: 10.1038/nature12509
|
| [15] |
Mosconi E, Amat A, Nazeeruddin M, et al. (2013) First-principles modeling of mixed halide organometal perovskites for photovoltaic applications. J Phys Chem C 117: 13902–13913. doi: 10.1021/jp4048659
|
| [16] |
Wang Y, Gould T, Dobson J, et al. (2014) Density functional theory analysis of structural and electronic properties of orthorhombic perovskite CH3NH3PbI3. Phys Chem Chem Phys 16: 1424–1429. doi: 10.1039/C3CP54479F
|
| [17] | Umari P, Mosconi E, De Angelis, F (2014) Relativistic GW calculations on CH3NH3PbI3 and CH3NH3SnI3 perovskites for solar cell applications. Sci Rep 4: Article number: 4467. |
| [18] |
Even J, Pedesseau L, Jancu J, et al. (2013) Importance of spin–orbit coupling in hybrid organic/inorganic perovskites for photovoltaic applications. J Phys Chem Lett 4: 2999–3005. doi: 10.1021/jz401532q
|
| [19] |
Even J, Pedesseau L, Dupertuis M, et al. (2012) Electronic model for self-assembled hybrid organic/perovskite semiconductors: reverse band edge electronic states ordering and spin-orbit coupling. Phys Rev B 86: 205301. doi: 10.1103/PhysRevB.86.205301
|
| [20] |
Even J, Pedesseau L, Katan C (2014) Comments on “density functional theory analysis of structural and electronic properties of orthorhombic perovskite CH3NH3PbI3.” Phys Chem Chem Phys 16:8697-8698 doi: 10.1039/C3CP55006K
|
| [21] |
Feng J, Xiao B (2014) Correction to “crystal structures, optical properties, and effective mass tensors of CH3NH3PbI3 (X=I and Br) phases predicted from HSE06.” J Phys Chem Lett 5: 1719-1720. doi: 10.1021/jz500831m
|
| [22] | Brivio F, Butler K, Walsh A (2014) Relativistic quasiparticle self-consistent electronic structure of hybrid halide perovskite photovoltaic absorbers. Phys Rev B 89: 155024 |
| [23] | Filippetti A, Mattoni A (2014) Hybrid perovskites for photovoltaics: insights from first principles. Phys Rev B 89: 125203 |
| [24] |
Jishi R, Ta O, Sharif A (2014) Modeling of lead halide compounds for photovoltaic applications. J Phys Chem C 118: 28344–28349. doi: 10.1021/jp5050145
|
| [25] |
Motta C, El-Mellouhi F, Kais S, et al. (2015) Revealing the role of organic cations in hybrid halide perovskite CH3NH3PbI3. Nat Commun 6: 7026. doi: 10.1038/ncomms8026
|
| [26] | Baikie T, Fang Y, Kadro J, et al. (2013) Synthesis and crystal chemistry of the hybrid perovskite (CH3NH3)PbI3 for solid-state sensitised solar cell applications. J Mater Chem A 1: 5628–5641. |
| [27] |
Comin R, Walters G, Thibau E, et al. (2015) Structural, optical, and electronic studies of widebandgap lead halide perovskites. J Mater Chem C 3: 8839–8843. doi: 10.1039/C5TC01718A
|
| [28] | Buin A, Comin R, Xu J, et al. (2015) Halide-dependent electronic structure of organolead perovskite materials. Chem Mater 27: 4405–4412. |
| [29] |
Pang S, Hu H, Zhang J, et al. (2014) NH2CH=NH2PbI3: An alternative organolead iodide perovskite sensitizer for mesoscopic solar cells. Chem Mater 26: 1485–1491. doi: 10.1021/cm404006p
|
| [30] | Stoumpos C, Malliakas C, Kanatzidis M (2013) Semiconducting tin and lead iodide perovskites with organic cations: phase transitions, high mobilities, and near-infrared photoluminescent properties. Inorg Chem 52: 9019–9038. |
| [31] | Stoumpos C, Kanatzidis G (2015) The renaissance of halide perovskites and their evolution as emerging semiconductors. Acc Chem Res 48: 2791–2802. |
| [32] |
Eperon G, Stranks S, Menelaou C, et al. (2014) Formamidinium lead trihalide: a broadly tunable perovskite for efficient planar heterojunction solar cells. Energy Environ Sci 7: 982–988. doi: 10.1039/c3ee43822h
|
| [33] |
Koh T, Fu K, Fang Y, et al. (2014) Formamidinium-containing metal halide: an alternative material for near-IR absorption perovskite solar cells. J Phys Chem C 118: 16458–16462. doi: 10.1021/jp411112k
|
| [34] |
Jeon N, Noh J, Yang W, et al. (2015) Compositional engineering of perovskite materials for highperformance solar cells. Nature 517: 476–480. doi: 10.1038/nature14133
|
| [35] |
Tan Z, Moghaddam R, Lai M, et al. (2014) Bright light-emitting diodes based on organometal halide perovskite. Nature Nanotech 9: 687–692. doi: 10.1038/nnano.2014.149
|
| [36] |
Kim Y.-H, Cho H, Heo J, et al. (2015) Multicolored organic/inorganic hybrid perovskite lightemitting diodes. Adv Mater 27: 1248–1254. doi: 10.1002/adma.201403751
|
| [37] |
Amat A, Mosconi E, Ronca E, et al. (2014) Cation-induced band-gap tuning in organohalide perovskites: interplay of spin-orbit coupling and octahedra tilting. Nano Lett 14: 3608–3616. doi: 10.1021/nl5012992
|
| [38] |
Kieslich G, Sun S, Cheetham A (2015) An extended tolerance factor approach for organic-inorganic perovskites. Chem Sci 6: 3430–3433. doi: 10.1039/C5SC00961H
|
| [39] | Mashiyama H, Kurihara Y, Azetsu T (1998) Disordered cubic perovskite structure of CH3NH3PbX3(X = Cl, Br, I). J Korean Phys Soc 32: S156-S158. |
| [40] | Becke A (1993) Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys 98: 5648–5652. |
| [41] | Frisch M, Trucks G, Schlegel H, et al. (2009) Gaussian 09, Gaussian, Inc: Willingford, CT. |
| [42] | Kohn W, Sham L (1965) Self-consistent equations including exchange and correlation effects. Phys Rev 140: A1133–A1138. |
| [43] | Perdew J, Burke K, Ernzerhof M (1996) Generalized gradient approximation made simple. Phys Rev Lett 77: 3865–3868. |
| [44] |
Bechstedt F, Fuchs F, Kresse G (2009) Ab-initio theory of semiconductor band structures: new developments and progress. Phys Status Solidi B 246: 1877–1892. doi: 10.1002/pssb.200945074
|
| [45] | Becke A, Johnson E (2006) A simple effective potential for exchange. J Chem Phys 124: 221101. |
| [46] | Tran F, Blaha P (2009) Accurate band gaps of semiconductors and insulators with a semilocal exchange-correlation potential. Phys Rev Lett 102: 226401. |
| [47] |
Becke A, Roussel M (1989) Exchange holes in inhomogeneous systems: a coordinate-space model. Phys Rev A 39: 3761–3767. doi: 10.1103/PhysRevA.39.3761
|
| [48] | Blaha P, Schwarz K, Madsen G, et al. (2001) WIEN2K: an augmented plane wave + local orbitals program for calculating crystal properties. |
| [49] |
Monkhorst H, Pack J (1976) Special points for Brillouin-zone integrations. Phys Rev B 13: 5188–5192. doi: 10.1103/PhysRevB.13.5188
|
| [50] |
Perdew J, Ruzsinzky A, Csonka G, et al. (2008) Restoring the density-gradient expansion for exchange in solids. Phys Rev Lett 100: 136406. doi: 10.1103/PhysRevLett.100.136406
|
| [51] | Wyckoff R (1963) Crystal structures, 2nd ed. (Wiley, New York) Vol. 1. |
| [52] |
Plekhanov V (2004) Lead halides: electronic properties and applications. Prog Mater Sci 49: 787–886. doi: 10.1016/S0079-6425(03)00049-5
|
| [53] |
Zaldo C, Sol´ e J, Di ´ eguez E, et al. (1985) Optical spectroscopy of PbCl2 particles embedded in NaCl host matrix. J Chem Phys 83: 6197–6200. doi: 10.1063/1.449859
|
| [54] |
Plekhanov V (1973) Optical constants of lead halides. Phys Stat Sol B 57: K55–K59. doi: 10.1002/pssb.2220570157
|
| [55] |
Iwanaga M, Watanabe M, Hayashi T (2000) Charge separation of excitons and the radiative recombination process in PbBr2 crystals. Phys Rev B 62: 10766–10773. doi: 10.1103/PhysRevB.62.10766
|
| [56] |
Matus M, Arduengo A, Dixon D (2006) The heats of formation of diazene, hydrazine, N2H3+, N2H5+, N2H, and N2H3 and the methyl derivatives CH3NNH, CH3NNCH3, and CH3HNNHCH3. J Phys Chem A 110: 10116–10121. doi: 10.1021/jp061854u
|
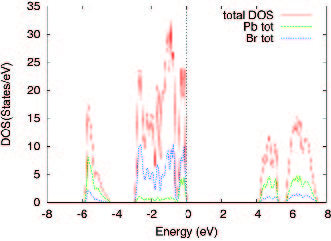
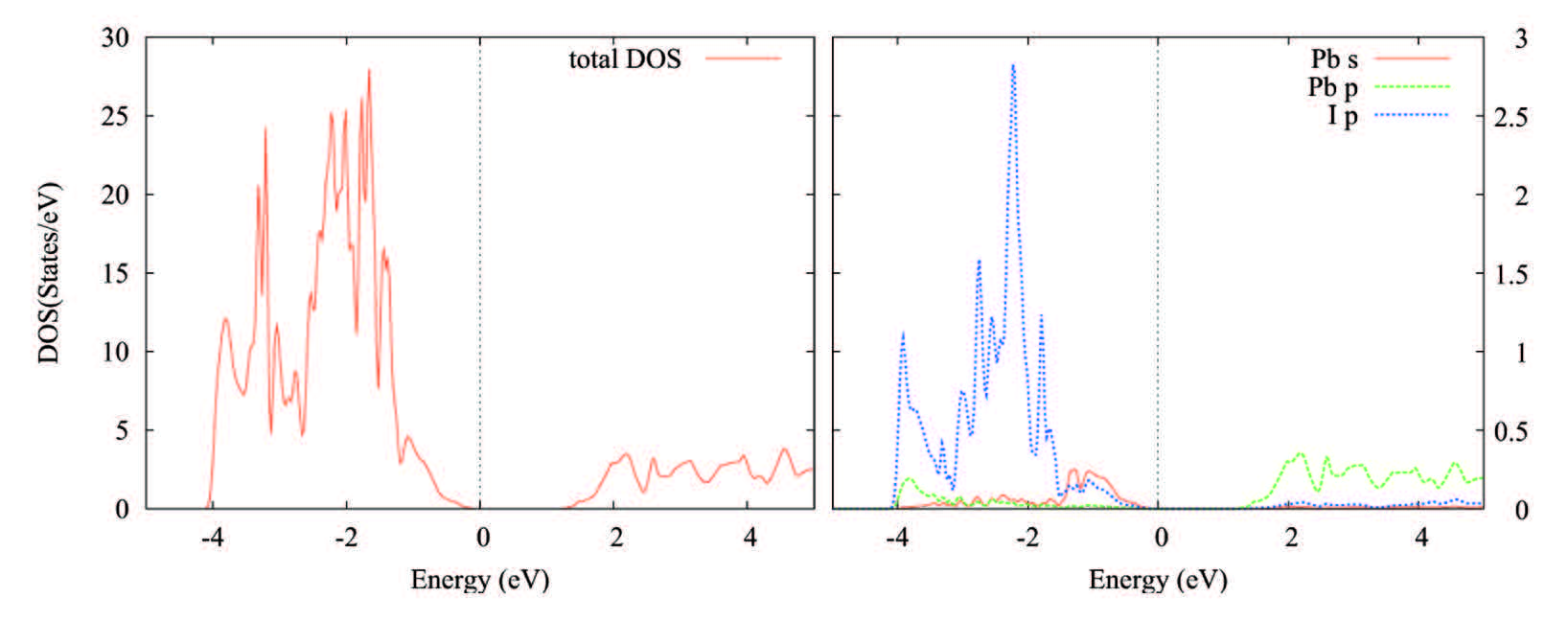
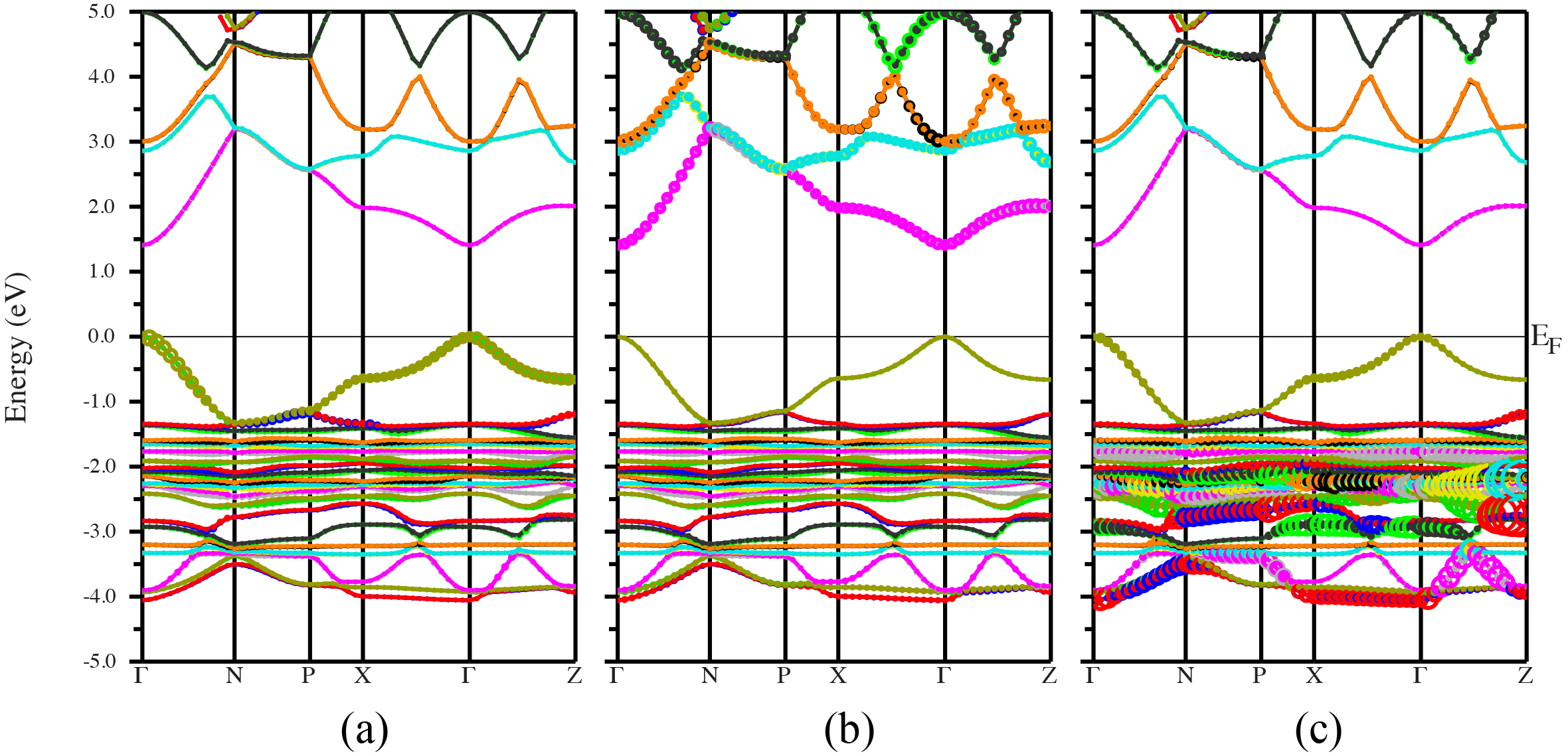
Figures(3) / Tables(3)

Radi A. Jishi. Modified Becke-Johnson exchange potential: improved modeling of lead halides for solar cell applications[J]. AIMS Materials Science, 2016, 3(1): 149-159. doi: 10.3934/matersci.2016.1.149







 DownLoad:
DownLoad: