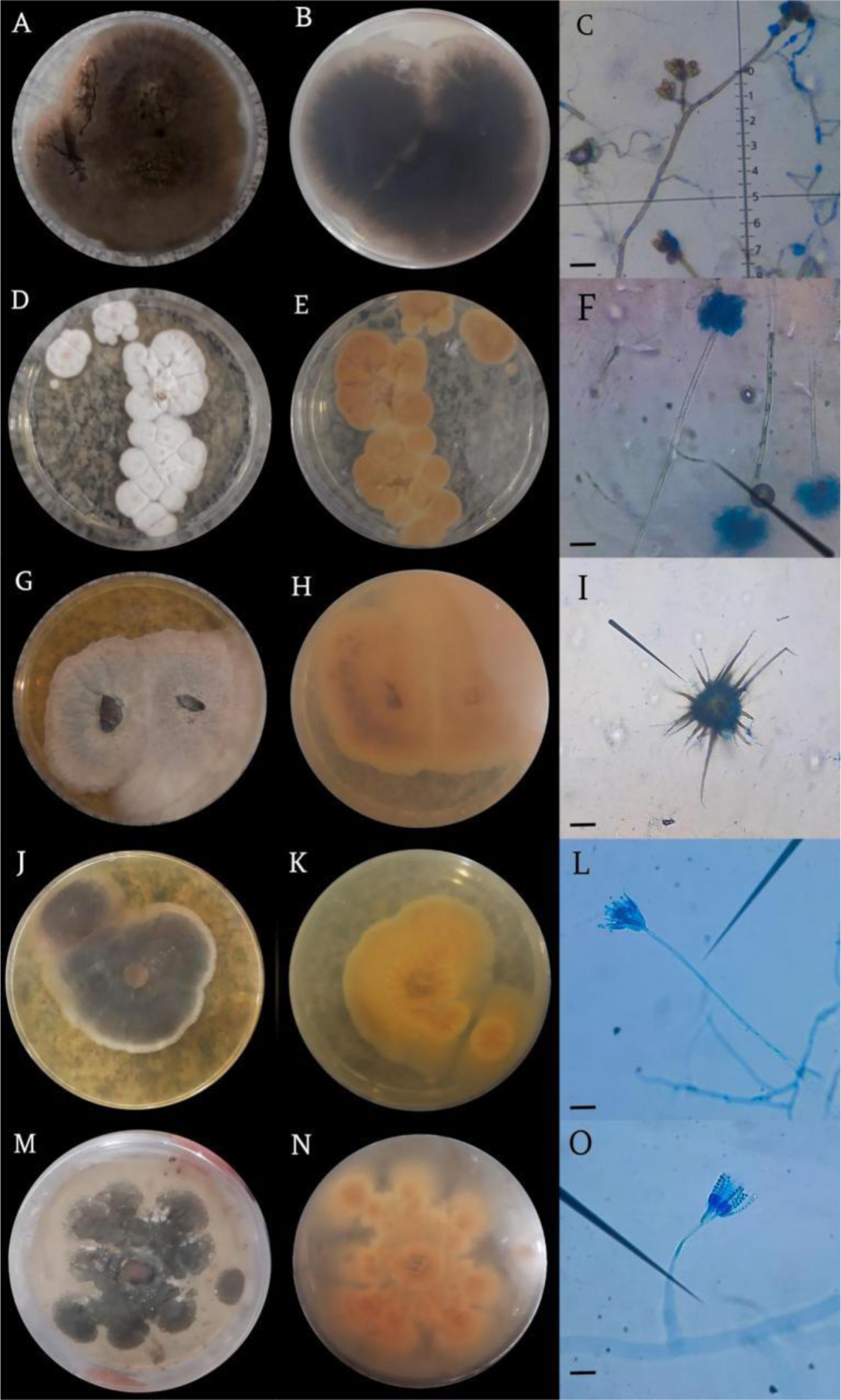
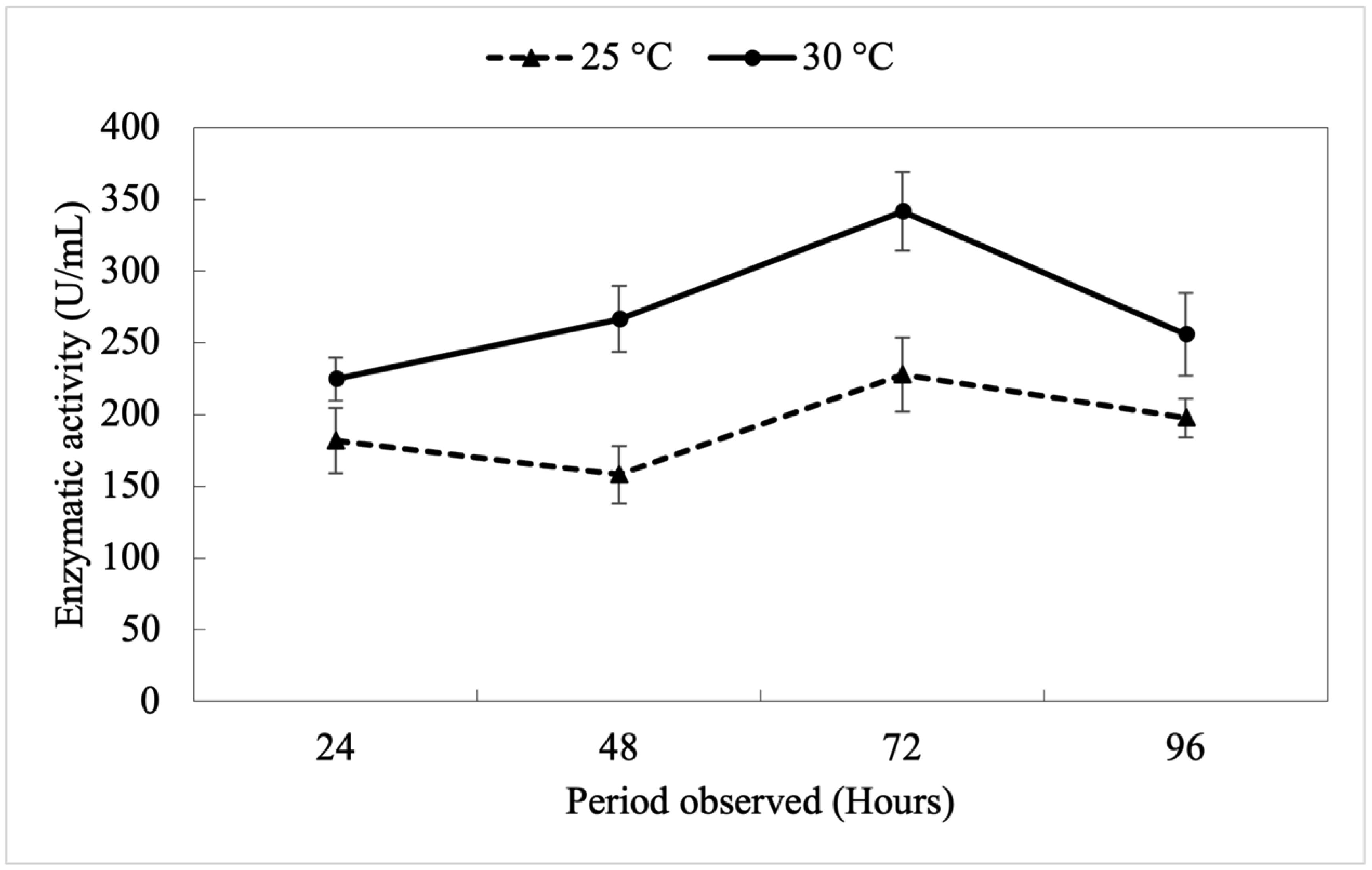
In this study, we focused on aquatic environments and explored the fungal biodiversity and protease production in the Tapajós River Basin in Pará, Brazil. Our objectives were to identify and characterize fungal genera with potential protease activity and evaluate their biotechnological applications. Variations in the enzymatic activity index, colony diameters, and halo sizes were documented. For molecular identification, genomic DNA was isolated and analyzed using both the ITS region and the 28S rRNA region, which provided additional resolution for the most promising isolate. Penicillium citrinum UFOPA-MI0019 exhibited significant protease activity, suggesting potential applications in biotechnological processes. We also included the kinetics of protease production of P. citrinum UFOPA-MI0019 under solid-state fermentation using wheat bran, indicating a peak (340 ± 30 UI/mL) in enzyme activity at 72 hours of fermentation at an optimal temperature of 30 °C. These findings highlight the ecological role and industrial potential of Amazonian fungi, contributing to sustainable waste transformation.
Citation: Aline Lima de Aguiar, Ana Luiza Figueira da Silva, Rayane Bonfim Ferreira Xavier, Marcos Diones Ferreira Santana, Dávia Marciana Talgatti, Fernando Abreu Oliveira, Carlos Ivan Aguillar-Vildoso, Érica Simplício de Souza, Márcio Barreto Rodrigues, João Paulo Alves Silva, Lívia Melo Carneiro, Clarice Maia Carvalho, João Vicente Braga de Souza, Eveleise Samira Martins Canto. Exploring the potential of fungal proteases in aquatic environments of the Tapajós Basin, Brazil[J]. AIMS Bioengineering, 2025, 12(1): 162-176. doi: 10.3934/bioeng.2025007
In this study, we focused on aquatic environments and explored the fungal biodiversity and protease production in the Tapajós River Basin in Pará, Brazil. Our objectives were to identify and characterize fungal genera with potential protease activity and evaluate their biotechnological applications. Variations in the enzymatic activity index, colony diameters, and halo sizes were documented. For molecular identification, genomic DNA was isolated and analyzed using both the ITS region and the 28S rRNA region, which provided additional resolution for the most promising isolate. Penicillium citrinum UFOPA-MI0019 exhibited significant protease activity, suggesting potential applications in biotechnological processes. We also included the kinetics of protease production of P. citrinum UFOPA-MI0019 under solid-state fermentation using wheat bran, indicating a peak (340 ± 30 UI/mL) in enzyme activity at 72 hours of fermentation at an optimal temperature of 30 °C. These findings highlight the ecological role and industrial potential of Amazonian fungi, contributing to sustainable waste transformation.

| [1] |
Abdullah AA, Babu J (2020) Current prospective in using cold-active enzymes as eco-friendly detergent additive. Appl Microbiol Biotechnol 104: 2871-2882. https://doi.org/10.1007/s00253-020-10429-x 
|
| [2] | Lima ADDR, Farias VA, Oliveira HD (2018) Patents technological prospecting of proteases used in cheese making. Prospect Noteb 11: 1726. https://doi.org/10.9771/cp.v12i5.27619 |
| [3] |
Cardoso KBB, Nascimento TP, Oliveira VM, et al. (2022) Protease with fibrinolytic and collagenolytic activity produced by Aspergillus ochraceus URM604. Res Soc Dev 11: e15511225500. https://doi.org/10.33448/rsd-v11i2.25500
|
| [4] | Climaco LR, Roder C, Olivo JE (2023) Production of proteases by fungi of the Aspergillus genus: the impact of cultivation conditions and their biotechnological implications. Obs Lat Am Econ 21: 17465-17481. https://doi.org/10.55905/oelv21n10-159 |
| [5] |
Paz Junior FB, Silva CRB, Freitas LR, et al. (2020) Evaluation of the proteolytic activity of Penicillium chrysogenum isolated from the IFPE - Campus Recife memorial room. Braz J Dev 6: 60502-60508. https://doi.org/10.34117/bjdv6n8-459
|
| [6] |
Santos BR, Maeda RN, Hamada N, et al. (2023) Screening of fungal strains isolated from Simuliidae (Insecta: Diptera) larvae to obtain enzymes of industrial interest in the Amazon. Braz J Anim Environ Res 6: 3350-3358. https://doi.org/10.34188/bjaerv6n4-021
|
| [7] |
Prado FB, Batista SCP, Martim SR, et al. (2021) Feasibility of protease production by Aspergillaceae species and screening of coagulants from bovine milk. Braz J Dev 7: 16356-16373. https://doi.org/10.34117/bjdv7n2-317
|
| [8] |
Oliveira Filho DC, Coelho KWSA, Costa VCA, et al. (2022) Synthesis of proteases by a species of anamorphic filamentous fungus for application in industrial processes. Res Soc Dev 11: e548111638526. https://doi.org/10.33448/rsd-v11i16.38526
|
| [9] | Canto ESM, Costa DVS, Sousa LS, et al. (2023) Evaluation of enzymatic activity production by filamentous fungi collected from Amazonian soil. Microb Biotechnol 1: 142-156. Available from: https://downloads.editoracientifica.com.br/articles/230111840.pdf |
| [10] |
Benchaya GA, França EM, Silva NCCA, et al. (2022) Proteases from a species of the Trichocomaceae family for application in the food industry. Res Soc Dev 11: e547111638525. https://doi.org/10.33448/rsd-v11i16.38525
|
| [11] |
Batista Junior GP, Silva KND, Santiago PAL, et al. (2021) Enzyme prospection and antimicrobial activity of Penicillium species isolated from the Amazon Biome. Braz J Dev 7: 75011-75025. https://doi.org/10.34117/bjdv7n7-603
|
| [12] |
Kieliszek M, Pobiega K, Piwowarek K, et al. (2021) Characteristics of the proteolytic enzymes produced by lactic acid bacteria. Molecules 26: 1858. https://doi.org/10.3390/molecules26071858
|
| [13] |
Santos AF, Parisi JJD, Menten JOM (2023) Diversity of endophytic filamentous fungi in seeds of native forest species in southern Brazil. Summa Phytopathol 49: e257357. https://doi.org/10.1590/0100-5405/257357
|
| [14] |
Tsang CC, Tang JY, Lau SK, et al. (2018) Taxonomy and evolution of Aspergillus, Penicillium and Talaromyces in the omics era–Past, present and future. Comput Struct Biotechnol J 16: 197-210. https://doi.org/10.1016/j.csbj.2018.05.003
|
| [15] | White TJ, Bruns T, Lee S, et al. (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR Protocols 18: 315-322. https://doi.org/10.1016/B978-0-12-372180-8.50042-1 |
| [16] | Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41: 95-98. |
| [17] |
Chen L, Daniel RM, Coolbear T (2003) Detection and impact of protease and lipase activities in milk and milk powders. Int Dairy J 13: 255-275. https://doi.org/10.1016/S0958-6946(02)00171-1
|
| [18] |
Florencio C, Couri S, Farinas, CS (2012) Correlation between agar plate screening and solid-state fermentation for the prediction of cellulase production by trichoderma strains. Enzyme Res 2012: 1-7. https://doi.org/10.1155/2012/793708
|
| [19] |
Merheb CW, Cabral H, Gomes E, et al. (2007) Partial characterization of protease from a thermophilic fungus, Thermoascus aurantiacus, and its hydrolytic activity on bovine casein. Food Chem 104: 127-131. https://doi.org/10.1016/j.foodchem.2006.11.010
|
| [20] |
Montoya S, Patiño A, Sánchez ÓJ (2021) Production of lignocellulolytic enzymes and biomass of Trametes versicolor from agro-industrial residues in a novel fixed-bed bioreactor with natural convection and forced aeration at pilot scale. Processes 9: 1-19. https://doi.org/10.3390/pr9020397
|
| [21] | Haq I, Mukhtar H, Umber H (2006) Production of protease by Penicillium chrysogenum through optimization of environmental conditions. J Agric Soc Sci 2: 23-25. https://www.researchgate.net/publication/238728994 |
| [22] |
Leighton TJ, Doi RH, Warren RAJ, et al. (1973) The relationship of serine protease activity to RNA polymerase modification and sporulation in Bacillus subtilis. J Mol Biol 76: 103-122. https://doi.org/10.1016/0022-2836(73)90083-1
|
| [23] |
Nicoletti R, Vinale F (2018) Bioactive compounds from marine-derived Aspergillus, Penicillium, Talaromyces and Trichoderma species. Mar Drugs 16: 408. https://doi.org/10.3390/md16110408
|
| [24] |
Khan NA, Asaf S, Ahmad W, et al. (2023) Diversity, lifestyle, genomics, and their functional role of Cochliobolus, Bipolaris, and Curvularia species in environmental remediation and plant growth promotion under biotic and abiotic stressors. J Fungi 9: 254. https://doi.org/10.3390%2Fjof9020254
|
| [25] |
Kaur M, Peshwani H, Goel M (2024) Penicillium: a treasure trove for antimycobacterial and antioxidant metabolites. Fungi Bioactive Metabolites: Integration of Pharmaceutical Applications . Singapore: Springer Nature Singapore 263-281. https://doi.org/10.1007/978-981-99-5696-8_9
|
| [26] |
Heo I, Hong K, Yang H, et al. (2019) Diversity of Aspergillus, Penicillium, and Talaromyces species isolated from freshwater environments in Korea. Mycobiology 47: 12-19. https://doi.org/10.1080/12298093.2019.1572262
|
| [27] |
Pangging M, Nguyen T, Lee H (2019) New records of four species belonging to Eurotiales from soil and freshwater in Korea. Mycobiology 47: 154-164. https://doi.org/10.1080/12298093.2018.1554777
|
| [28] |
You YH, Park JM, Park JH, et al. (2015) Diversity of endophytic fungi associated with the roots of four aquatic plants inhabiting two wetlands in Korea. Mycobiology 43: 231-238. https://doi.org/10.5941/MYCO.2015.43.3.231
|
| [29] |
Lücking R, Aime MC, Robbertse B, et al. (2020) Unambiguous identification of fungi: where do we stand and how accurate and precise is fungal DNA barcoding?. IMA Fungus 11: 1-32. https://doi.org/10.1186/s43008-020-00033-z
|
| [30] |
Queiroz C, Sousa ACB (2020) Production of hydrolytic enzymes by filament fungi in different solid substrates. Braz J Dev 6: 51849-51860. https://doi.org/10.34117/bjdv6n7-725
|
| [31] | Ikhsanudin A, Emantis R, Ekowati CN (2019) Proteolytic activity of the entomopathogenic fungi (Penicillium sp.) of cockroaches (Periplaneta americana). J IIm Biol EKspe Keanekaragaman Hayati 6: 81-84. https://doi.org/10.23960/jbekh.v6i2.47 |
| [32] |
Harwood CR, Kikuchi Y (2022) The ins and outs of Bacillus proteases: activities, functions and commercial significance. FEMS Microbiol Rev 46: 1-20. https://doi.org/10.1093/femsre/fuab046
|
| [33] | Sethi S, Gupta S Optimization of protease production from fungi isolated from soil (2015). Available from: https://pesquisa.bvsalud.org/portal/resource/pt/sea-168920 |
| [34] |
Behera BC, Bijay KS, Mohapatra S, et al. (2021) Bio-production of alkaline protease by Trichoderma longibrachiatum and Penicillium rubidurum using different agro-industrial products. Novel Res Microbiol J 5: 1241-1255. https://doi.org/10.21608/nrmj.2021.178300
|
| [35] | Chapla VM, Biasetto CR, Araujo AR (2013) Endophytic Fungi: An unexplored and sustainable source of new and bioactive natural products. Virtual Quim 5: 421-437. https://doi.org/10.5935/1984-6835.20130036 |
| [36] |
Bogas AC, Cruz FPN, Lacava PT, et al. (2022) Endophytic fungi: an overview on biotechnological and agronomic potential. Braz J Biol 84: e258557. https://doi.org/10.1590/1519-6984.258557
|
| [37] |
Guillaume A, Thorigné A, Carré Y, et al. (2019) Contribution of proteases and cellulases produced by solid-state fermentation to the improvement of corn ethanol production. Bioresour Bioprocess 6: 1-12. https://doi.org/10.1186/s40643-019-0241-0
|
| [38] |
Figueiredo CCM, Granero FO, Silva LP, et al. (2024) Solid-state fermentation using wheat bran to produce glucose syrup and functional cereal bars. Bioprocess Biosyst Eng 47: 1081-1094. https://doi.org/10.1007/s00449-024-03032-1
|
| [39] | Freire AFN, Canto ESM, Paiva MJM, et al. (2024) Scientific and technological prospecting of environmental yeasts regarding the production of hydrolytic enzymes of biotechnological interest. Contrib Cienc Soc 17: 1499-1512. https://doi.org/10.55905/revconv.17n.1-084 |
Figures(2) / Tables(2)

Aline Lima de Aguiar, Ana Luiza Figueira da Silva, Rayane Bonfim Ferreira Xavier, Marcos Diones Ferreira Santana, Dávia Marciana Talgatti, Fernando Abreu Oliveira, Carlos Ivan Aguillar-Vildoso, Érica Simplício de Souza, Márcio Barreto Rodrigues, João Paulo Alves Silva, Lívia Melo Carneiro, Clarice Maia Carvalho, João Vicente Braga de Souza, Eveleise Samira Martins Canto. Exploring the potential of fungal proteases in aquatic environments of the Tapajós Basin, Brazil[J]. AIMS Bioengineering, 2025, 12(1): 162-176. doi: 10.3934/bioeng.2025007






 DownLoad:
DownLoad: