Research article
Special Issues
Can the external masculinization score predict the success of genetic testing in 46,XY DSD?
-
1.
Department of Urology, Division of Pediatric Urology, University of Wisconsin School of Medicine and Public Health, 1685 Highland Avenue, Madison, Wisconsin 53705, USA;
-
2.
Department of Pediatrics, Division of Genetic Medicine, University of Washington School of Medicine, 4800 Sandpoint Way NE, Seattle, WA 98105, USA;
-
3.
Department of Pediatrics, Division of Endocrinology, University of Washington School of Medicine, 4800 Sandpoint Way NE, Seattle, WA 98105, USA;
-
4.
Department of Urology, Division of Pediatric Urology, University of Washington School of Medicine, 4800 Sandpoint Way NE, Seattle, WA 98105, USA
-
Received:
29 November 2014
Accepted:
27 April 2015
Published:
07 May 2015
-
-
-
-
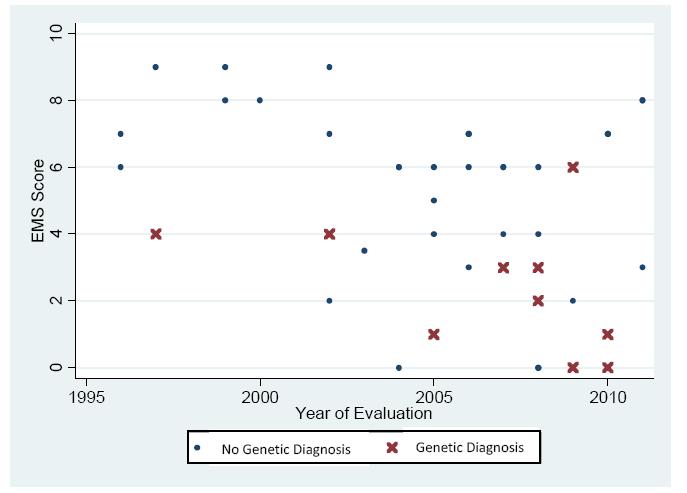
Genetic testing is judiciously applied to individuals with Disorders of Sex Development (DSD) and so it is necessary to identify those most likely to benefit from such testing. We hypothesized that the external masculinization score (EMS) is inversely associated with the likelihood of finding a pathogenic genetic variant. Patients with 46,XY DSD from a single institution evaluated from 1994-2014 were included. Results of advanced cytogenetic and gene sequencing tests were recorded. An EMS score (range 0-12) was assigned to each patient according to the team's initial external genitalia physical examination. During 1994-2011, 44 (40%) patients with 46,XY DSD were evaluated and underwent genetic testing beyond initial karyotype; 23% (10/44) had a genetic diagnosis made by gene sequencing or array. The median EMS score of those with an identified pathogenic variant was significantly different from those in whom no confirmed genetic cause was identified [median 3 (95% CI, 2-6) versus 6 (95% CI, 5-7), respectively (p = 0.02)], but limited to diagnoses of complete or partial androgen insensitivity (8/10) or 5-reductase deficiency (2/10). In the modern cohort (2012-2014), the difference in median EMS in whom a genetic cause was or was not identified approached significance (p = 0.05, median 3 (95% CI, 0-7) versus 7 (95% CI, 6-9), respectively). When all patients from 1994-2014 are pooled, the EMS is significantly different amongst those with compared to those without a genetic cause (median EMS 3 vs. 6, p < 0.02). We conclude that an EMS of 3 or less may indicate a higher likelihood of identifying a genetic cause of 46,XY DSD and justify genetic screening, especially when androgen insensitivity is suspected.
Citation: Ruthie Su, Margaret P. Adam, Linda Ramsdell, Patricia Y. Fechner, Margarett Shnorhavorian. Can the external masculinization score predict the success of genetic testing in 46,XY DSD?[J]. AIMS Genetics, 2015, 2(2): 163-172. doi: 10.3934/genet.2015.2.163
-
Abstract
Genetic testing is judiciously applied to individuals with Disorders of Sex Development (DSD) and so it is necessary to identify those most likely to benefit from such testing. We hypothesized that the external masculinization score (EMS) is inversely associated with the likelihood of finding a pathogenic genetic variant. Patients with 46,XY DSD from a single institution evaluated from 1994-2014 were included. Results of advanced cytogenetic and gene sequencing tests were recorded. An EMS score (range 0-12) was assigned to each patient according to the team's initial external genitalia physical examination. During 1994-2011, 44 (40%) patients with 46,XY DSD were evaluated and underwent genetic testing beyond initial karyotype; 23% (10/44) had a genetic diagnosis made by gene sequencing or array. The median EMS score of those with an identified pathogenic variant was significantly different from those in whom no confirmed genetic cause was identified [median 3 (95% CI, 2-6) versus 6 (95% CI, 5-7), respectively (p = 0.02)], but limited to diagnoses of complete or partial androgen insensitivity (8/10) or 5-reductase deficiency (2/10). In the modern cohort (2012-2014), the difference in median EMS in whom a genetic cause was or was not identified approached significance (p = 0.05, median 3 (95% CI, 0-7) versus 7 (95% CI, 6-9), respectively). When all patients from 1994-2014 are pooled, the EMS is significantly different amongst those with compared to those without a genetic cause (median EMS 3 vs. 6, p < 0.02). We conclude that an EMS of 3 or less may indicate a higher likelihood of identifying a genetic cause of 46,XY DSD and justify genetic screening, especially when androgen insensitivity is suspected.

References
|
[1]
|
Baxter RM, Vilain E (2013) Translational genetics for diagnosis of human disorders of sex development. Annu Rev Genomics Hum Genet 14: 371-392. doi: 10.1146/annurev-genom-091212-153417

|
|
[2]
|
Hughes IA, Houk C, Ahmed SF, et al. (2006) Consensus statement on management of intersex disorders. Arch Dis Child 91: 554-563.
|
|
[3]
|
Ahmed SF, Khwaja O, Hughes IA (2000) The role of a clinical score in the assessment of ambiguous genitalia. BJU Int 85: 120-124. doi: 10.1046/j.1464-410x.2000.00354.x
|
|
[4]
|
Barthold JS (2011) Disorders of sex differentiation: a pediatric urologist's perspective of new terminology and recommendations. J Urol 185: 393-400. doi: 10.1016/j.juro.2010.09.083
|
|
[5]
|
Palmer JS (2012) Abnormalities of the External Genitalia in Boys. Campbell-Walsh Urology. 10th ed. 3537-3556.
|
|
[6]
|
Ono M, Harley VR (2013) Disorders of sex development: new genes, new concepts. Nat Rev Endocrinol 9: 79-91.
|
|
[7]
|
Deeb A, Mason C, Lee YS, et al. (2005) Correlation between genotype, phenotype and sex of rearing in 111 patients with partial androgen insensitivity syndrome. Clin Endocrinol (Oxf) 63:56-62. doi: 10.1111/j.1365-2265.2005.02298.x
|
|
[8]
|
Cools M, Pleskacova J, Stoop H, et al. (2011) Gonadal pathology and tumor risk in relation to clinical characteristics in patients with 45,X/46,XY mosaicism. J Clin Endocrinol Metab 96: E1171-1180. doi: 10.1210/jc.2011-0232
|
|
[9]
|
Baetens D, Mladenov W, Delle Chiaie B, et al. (2014) Extensive clinical, hormonal and genetic screening in a large consecutive series of 46, XY neonates and infants with atypical sexual development. Orphanet J Rare Dis 9: 209. doi: 10.1186/s13023-014-0209-2
|
|
[10]
|
Ahmed SF, Cheng A, Dovey L, et al. (2000) Phenotypic features, androgen receptor binding, and mutational analysis in 278 clinical cases reported as androgen insensitivity syndrome. J Clin Endocrinol Metab 85: 658-665.
|
-
-
-
-








 DownLoad:
DownLoad: