Citation: Carla Marchetti. Green tea catechins and intracellular calcium dynamics in prostate cancer cells[J]. AIMS Molecular Science, 2021, 8(1): 1-12. doi: 10.3934/molsci.2021001

| [1] |
Berridge MJ, Bootman MD, Lipp P (1998) Calcium - A life and death signal. Nature 395: 645-648. doi: 10.1038/27094

|
| [2] |
Bootman MD, Bultynck G (2020) Fundamentals of cellular calcium signaling: A primer. Cold Spring Harb Perspect Biol 12: 1-16. doi: 10.1101/cshperspect.a038802
|
| [3] |
Prevarskaya N, Skryma R, Shuba Y (2004) Ca2+ homeostasis in apoptotic resistance of prostate cancer cells. Biochem Biophys Res Commun 322: 1326-1335. doi: 10.1016/j.bbrc.2004.08.037
|
| [4] |
Monteith GR, Prevarskaya N, Roberts-Thomson SJ (2017) The calcium-cancer signalling nexus. Nat Rev Cancer 17: 367-380. doi: 10.1038/nrc.2017.18
|
| [5] |
Vanoverberghe K, Vanden Abeele F, Mariot P, et al. (2004) Ca2+ homeostasis and apoptotic resistance of neuroendocrine-differentiated prostate cancer cells. Cell Death Differ 11: 321-330. doi: 10.1038/sj.cdd.4401375
|
| [6] |
Kucera R, Pecen L, Topolcan O, et al. (2020) Prostate cancer management: long-term beliefs, epidemic developments in the early twenty-first century and 3PM dimensional solutions. EPMA J 11: 399-418. doi: 10.1007/s13167-020-00214-1
|
| [7] |
Siegel RL, Miller KD, Jemal A (2020) Cancer statistics, 2020. CA Cancer J Clin 70: 7-30. doi: 10.3322/caac.21590
|
| [8] |
Saraon P, Jarvi K, Diamandis EP (2011) Molecular Alterations during Progression of Prostate Cancer to Androgen Independence. Clin Chem 57: 1366-1375. doi: 10.1373/clinchem.2011.165977
|
| [9] |
Howard N, Clementino M, Kim D, et al. (2019) New developments in mechanisms of prostate cancer progression. Semin Cancer Biol 57: 111-116. doi: 10.1016/j.semcancer.2018.09.003
|
| [10] |
Boutin B, Tajeddine N, Monaco G, et al. (2015) Endoplasmic reticulum Ca2+ content decrease by PKA-dependent hyperphosphorylation of type 1 IP3 receptor contributes to prostate cancer cell resistance to androgen deprivation. Cell Calcium 57: 312-320. doi: 10.1016/j.ceca.2015.02.004
|
| [11] |
Flourakis M, Lehen'kyi V, Beck B, et al. (2010) Orai1 contributes to the establishment of an apoptosis-resistant phenotype in prostate cancer cells. Cell Death Dis 1: e75. doi: 10.1038/cddis.2010.52
|
| [12] |
Dubois C, Vanden Abeele F, Lehen'kyi V, et al. (2014) Remodeling of Channel-Forming ORAI Proteins Determines an Oncogenic Switch in Prostate Cancer. Cancer Cell 26: 19-32. doi: 10.1016/j.ccr.2014.04.025
|
| [13] |
Perrouin Verbe MA, Bruyere F, Rozet F, et al. (2016) Expression of store-operated channel components in prostate cancer: The prognostic paradox. Hum Pathol 49: 77-82. doi: 10.1016/j.humpath.2015.09.042
|
| [14] |
Kampa M, Nifli AP, Notas G, et al. (2007) Polyphenols and cancer cell growth. Rev Physiol Biochem Pharmacol 159: 79-113. doi: 10.1007/112_2006_0702
|
| [15] |
Jian L, Xie LP, Lee AH, et al. (2004) Protective effect of green tea against prostate cancer: A case-control study in southeast China. Int J Cancer 108: 130-135. doi: 10.1002/ijc.11550
|
| [16] |
Bettuzzi S, Brausi M, Rizzi F, et al. (2006) Chemoprevention of human prostate cancer by oral administration of green tea catechins in volunteers with high-grade prostate intraepithelial neoplasia: A preliminary report from a one-year proof-of-principle study. Cancer Res 66: 1234-1240. doi: 10.1158/0008-5472.CAN-05-1145
|
| [17] |
Brausi M, Rizzi F, Bettuzzi S (2008) Chemoprevention of Human Prostate Cancer by Green Tea Catechins: Two Years Later. A Follow-up Update. Eur Urol 54: 472-473. doi: 10.1016/j.eururo.2008.03.100
|
| [18] |
Yuan JM (2013) Cancer prevention by green tea: evidence from epidemiologic studies. Am J Clin Nutr 98: 1676S-1681S. doi: 10.3945/ajcn.113.058271
|
| [19] | Perletti G, Magri V, Vral A, et al. (2019) Green tea catechins for chemoprevention of prostate cancer in patients with histologically-proven HG-PIN or ASAP. Concise review and meta-analysis. Arch Ital di Urol e Androl 91: 153-156. |
| [20] |
Cui K, Li X, Du Y, et al. (2017) Chemoprevention of prostate cancer in men with high-grade prostatic intraepithelial neoplasia (HGPIN): a systematic review and adjusted indirect treatment comparison. Oncotarget 8: 36674-36684. doi: 10.18632/oncotarget.16230
|
| [21] |
Guo Y, Zhi F, Chen P, et al. (2017) Green tea and the risk of prostate cancer. Medicine (Baltimore) 96: e6426. doi: 10.1097/MD.0000000000006426
|
| [22] |
Kurahashi N, Sasazuki S, Iwasaki M, et al. (2008) Green tea consumption and prostate cancer risk in Japanese men: A prospective study. Am J Epidemiol 167: 71-77. doi: 10.1093/aje/kwm249
|
| [23] | Filippini T, Malavolti M, Borrelli F, et al. (2020) Green tea (Camellia sinensis) for the prevention of cancer. Cochrane Database Syst Rev 3: CD005004. |
| [24] |
Tauber AL, Schweiker SS, Levonis SM (2020) From tea to treatment; epigallocatechin gallate and its potential involvement in minimizing the metabolic changes in cancer. Nutr Res 74: 23-36. doi: 10.1016/j.nutres.2019.12.004
|
| [25] |
Miyata Y, Shida Y, Hakariya T, et al. (2019) Anti-cancer effects of green tea polyphenols against prostate cancer. Molecules 24: 193-212. doi: 10.3390/molecules24010193
|
| [26] |
Sang S, Lambert JD, Ho CT, et al. (2011) The chemistry and biotransformation of tea constituents. Pharmacol Res 64: 87-99. doi: 10.1016/j.phrs.2011.02.007
|
| [27] |
Gan RY, Li HB, Sui ZQ, et al. (2018) Absorption, metabolism, anti-cancer effect and molecular targets of epigallocatechin gallate (EGCG): An updated review. Crit Rev Food Sci Nutr 58: 924-941. doi: 10.1080/10408398.2016.1231168
|
| [28] |
Albrecht DS, Clubbs EA, Ferruzzi M, et al. (2008) Epigallocatechin-3-gallate (EGCG) inhibits PC-3 prostate cancer cell proliferation via MEK-independent ERK1/2 activation. Chem Biol Interact 171: 89-95. doi: 10.1016/j.cbi.2007.09.001
|
| [29] |
Gupta S, Hussain T, Mukhtar H (2003) Molecular pathway for (−)-epigallocatechin-3-gallate-induced cell cycle arrest and apoptosis of human prostate carcinoma cells. Arch Biochem Biophys 410: 177-185. doi: 10.1016/S0003-9861(02)00668-9
|
| [30] |
Hagen RM, Chedea VS, Mintoff CP, et al. (2013) Epigallocatechin-3-gallate promotes apoptosis and expression of the caspase 9a splice variant in PC3 prostate cancer cells. Int J Oncol 43: 194-200. doi: 10.3892/ijo.2013.1920
|
| [31] |
Ju J, Lu G, Lambert JD, et al. (2007) Inhibition of carcinogenesis by tea constituents. Semin Cancer Biol 17: 395-402. doi: 10.1016/j.semcancer.2007.06.013
|
| [32] |
Yang CS, Wang H, Chen JX, et al. (2014) Effects of Tea Catechins on Cancer Signaling Pathways. Enzymes Academic Press, 195-221. doi: 10.1016/B978-0-12-802215-3.00010-0
|
| [33] |
Kim HS, Quon MJ, Kim JA (2014) New insights into the mechanisms of polyphenols beyond antioxidant properties; lessons from the green tea polyphenol, epigallocatechin 3-gallate. Redox Biol 2: 187-195. doi: 10.1016/j.redox.2013.12.022
|
| [34] |
Hastak K, Agarwal MK, Mukhtar H, et al. (2005) Ablation of either p21 or Bax prevents p53-dependent apoptosis induced by green tea polyphenol epigallocatechin-3-gallate. FASEB J 19: 1-19. doi: 10.1096/fj.04-2226fje
|
| [35] |
Van Aller GS, Carson JD, Tang W, et al. (2011) Epigallocatechin gallate (EGCG), a major component of green tea, is a dual phosphoinositide-3-kinase/mTOR inhibitor. Biochem Biophys Res Commun 406: 194-199. doi: 10.1016/j.bbrc.2011.02.010
|
| [36] |
Siddiqui IA, Asim M, Hafeez BB, et al. (2011) Green tea polyphenol EGCG blunts androgen receptor function in prostate cancer. FASEB J 25: 1198-1207. doi: 10.1096/fj.10-167924
|
| [37] |
Morré DJ, Morré DM, Sun H, et al. (2003) Tea Catechin Synergies in Inhibition of Cancer Cell Proliferation and of a Cancer Specific Cell Surface Oxidase (ECTO-NOX). Pharmacol Toxicol 92: 234-241. doi: 10.1034/j.1600-0773.2003.920506.x
|
| [38] |
Marchetti C, Gavazzo P, Burlando B (2020) Epigallocatechin-3-gallate mobilizes intracellular Ca2+ in prostate cancer cells through combined Ca2+ entry and Ca2+-induced Ca2+ release. Life Sci 258: 118232. doi: 10.1016/j.lfs.2020.118232
|
| [39] |
Van Bokhoven A, Varella-Garcia M, Korch C, et al. (2003) Molecular Characterization of Human Prostate Carcinoma Cell Lines. Prostate 57: 205-225. doi: 10.1002/pros.10290
|
| [40] |
Tai S, Sun Y, Squires JM, et al. (2011) PC3 is a cell line characteristic of prostatic small cell carcinoma. Prostate 71: 1668-1679. doi: 10.1002/pros.21383
|
| [41] | Grynkiewicz G, Poenie M, Tsien RY (1985) A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem 25: 3440-3450. |
| [42] |
Friedman M, Mackey BE, Kim HJ, et al. (2007) Structure-activity relationships of tea compounds against human cancer cells. J Agric Food Chem 55: 243-253. doi: 10.1021/jf062276h
|
| [43] |
Lewandowska U, Gorlach S, Owczarek K, et al. (2014) Synergistic interactions between anticancer chemotherapeutics and phenolic compounds and anticancer synergy between polyphenols. Postepy Hig Med Dosw 68: 528-540. doi: 10.5604/17322693.1102278
|
| [44] |
Kennedy DO, Matsumoto M, Kojima A, et al. (1999) Cellular thiols status and cell death in the effect of green tea polyphenols in Ehrlich ascites tumor cells. Chem Biol Interact 122: 59-71. doi: 10.1016/S0009-2797(99)00114-3
|
| [45] |
Clapham DE, Runnels LW, Strübing C (2001) The trp ion channel family. Nat Rev Neurosci 2: 387-396. doi: 10.1038/35077544
|
| [46] |
Thebault S, Roudbaraki M, Sydorenko V, et al. (2003) α1-adrenergic receptors activate Ca2+-permeable cationic channels in prostate cancer epithelial cells. J Clin Invest 111: 1691-1701. doi: 10.1172/JCI16293
|
| [47] |
Miller M, Shi J, Zhu Y, et al. (2011) Identification of ML204, a Novel Potent Antagonist That Selectively Modulates Native TRPC4/C5 Ion Channels. J Biol Chem 286: 33436-33446. doi: 10.1074/jbc.M111.274167
|
| [48] |
Sun YH, Gao X, Tang YJ, et al. (2006) Androgens induce increases in intracellular calcium via a G protein-coupled receptor in LNCaP prostate cancer cells. J Androl 27: 671-678. doi: 10.2164/jandrol.106.000554
|
| [49] | Loughlin KR (2014) Calcium channel blockers and prostate cancer. Urol Oncol Semin Orig Investig 32: 537-538. |
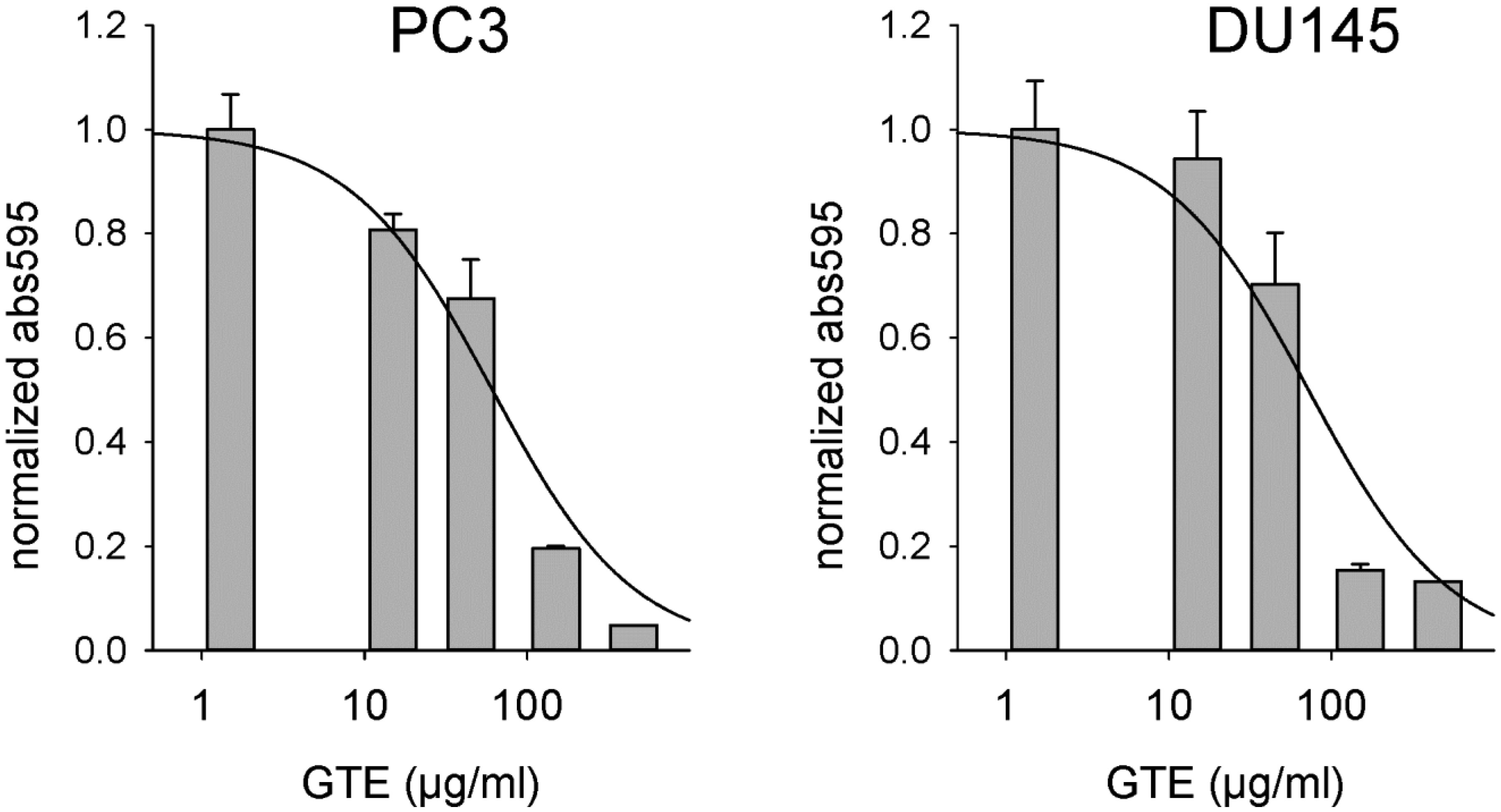
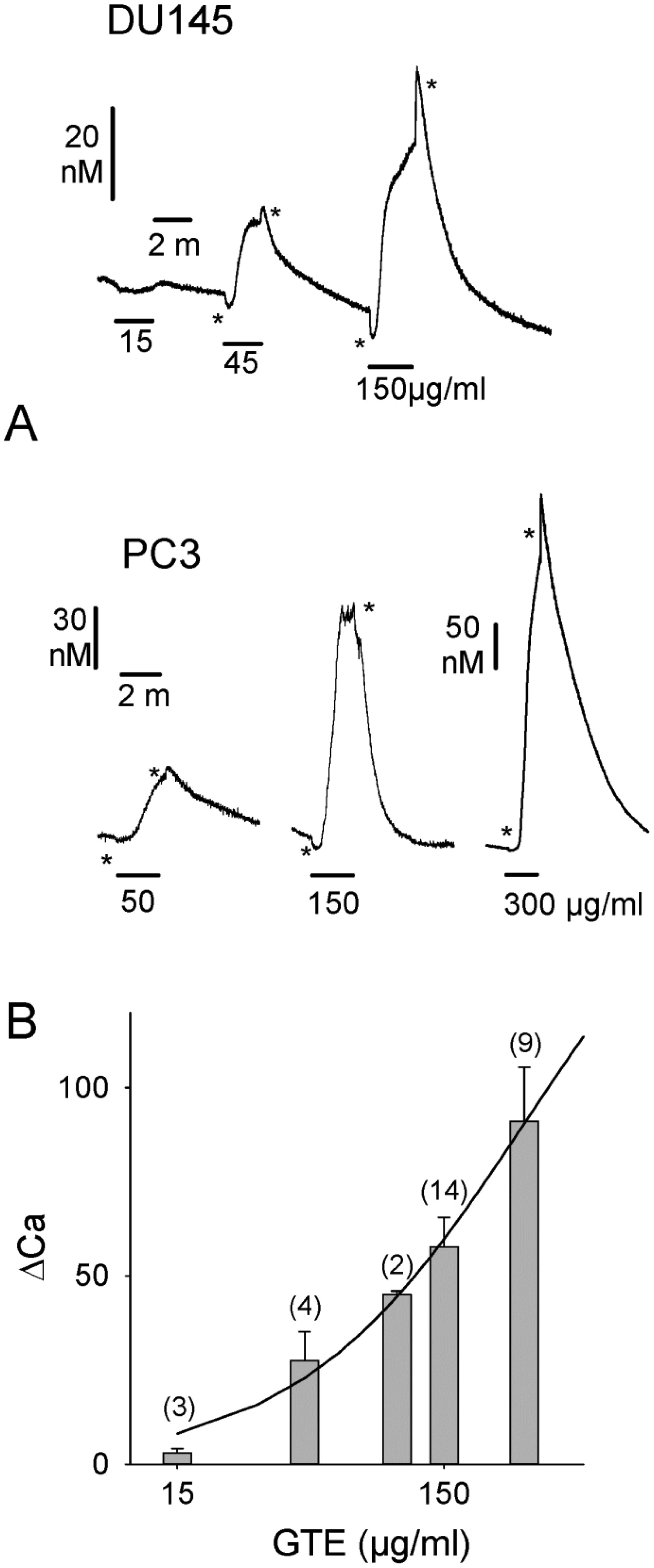
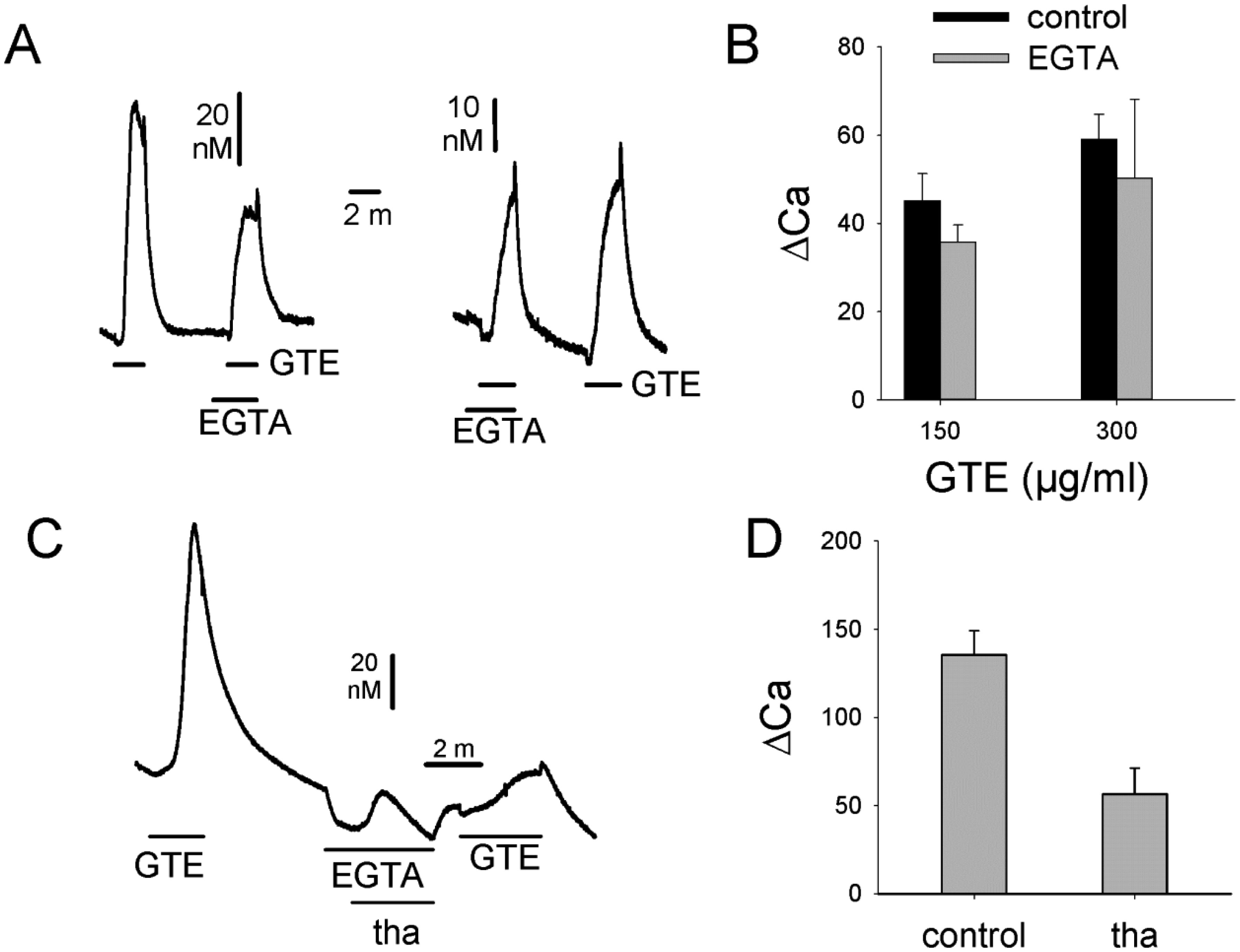
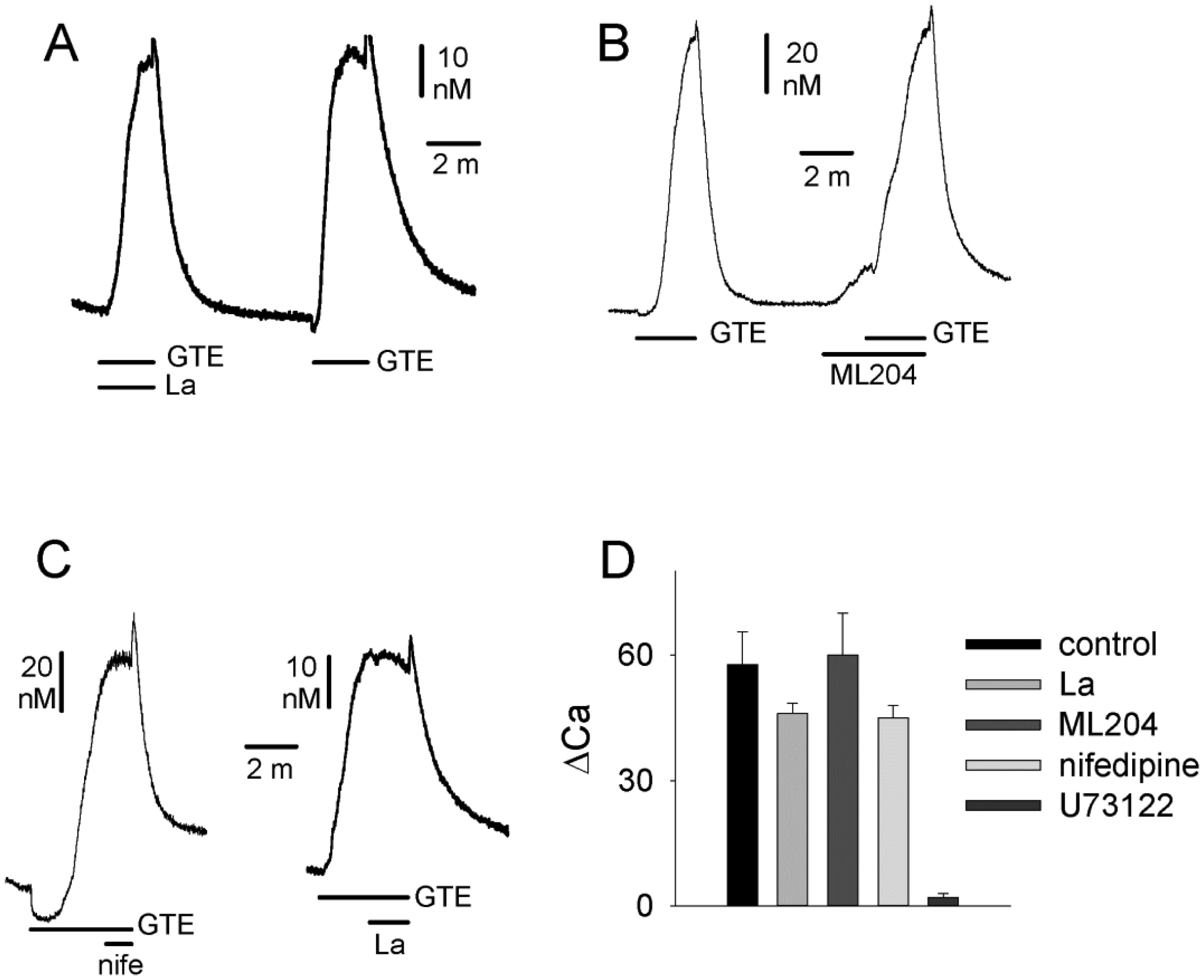
Figures(4)

Carla Marchetti. Green tea catechins and intracellular calcium dynamics in prostate cancer cells[J]. AIMS Molecular Science, 2021, 8(1): 1-12. doi: 10.3934/molsci.2021001







 DownLoad:
DownLoad: