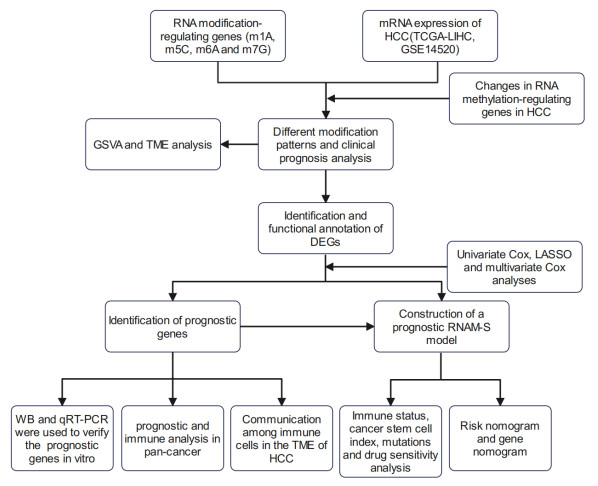
Background: Multiple types of RNA modifications are associated with the prognosis of hepatocellular carcinoma (HCC) patients. However, the overall mediating effect of RNA modifications on the tumor microenvironment (TME) and the prognosis of patients with HCC is unclear. Methods: Thoroughly analyze the TME, biological processes, immune infiltration and patient prognosis based on RNA modification patterns and gene patterns. Construct a prognostic model (RNA modification score, RNAM-S) to predict the overall survival (OS) in HCC patients. Analyze the immune status, cancer stem cell (CSC), mutations and drug sensitivity of HCC patients in both the high and low RNAM-S groups. Verify the expression levels of the four characteristic genes of the prognostic RNAM-S using in vitro cell experiments. Results: Two modification patterns and two gene patterns were identified in this study. Both the high-expression modification pattern and the gene pattern exhibited worse OS. A prognostic RNAM-S model was constructed based on four featured genes (KIF20A, NR1I2, NR2F1 and PLOD2). Cellular experiments suggested significant dysregulation of the expression levels of these four genes. In addition, validation of the RNAM-S model using each data set showed good predictive performance of the model. The two groups of HCC patients (high and low RNAM-S groups) exhibited significant differences in immune status, CSC, mutation and drug sensitivity. Conclusion: The findings of the study demonstrate the clinical value of RNA modifications, which provide new insights into the individualized treatment for patients with HCC.
Citation: Yuanqian Yao, Jianlin Lv, Guangyao Wang, Xiaohua Hong. Multi-omics analysis and validation of the tumor microenvironment of hepatocellular carcinoma under RNA modification patterns[J]. Mathematical Biosciences and Engineering, 2023, 20(10): 18318-18344. doi: 10.3934/mbe.2023814
Background: Multiple types of RNA modifications are associated with the prognosis of hepatocellular carcinoma (HCC) patients. However, the overall mediating effect of RNA modifications on the tumor microenvironment (TME) and the prognosis of patients with HCC is unclear. Methods: Thoroughly analyze the TME, biological processes, immune infiltration and patient prognosis based on RNA modification patterns and gene patterns. Construct a prognostic model (RNA modification score, RNAM-S) to predict the overall survival (OS) in HCC patients. Analyze the immune status, cancer stem cell (CSC), mutations and drug sensitivity of HCC patients in both the high and low RNAM-S groups. Verify the expression levels of the four characteristic genes of the prognostic RNAM-S using in vitro cell experiments. Results: Two modification patterns and two gene patterns were identified in this study. Both the high-expression modification pattern and the gene pattern exhibited worse OS. A prognostic RNAM-S model was constructed based on four featured genes (KIF20A, NR1I2, NR2F1 and PLOD2). Cellular experiments suggested significant dysregulation of the expression levels of these four genes. In addition, validation of the RNAM-S model using each data set showed good predictive performance of the model. The two groups of HCC patients (high and low RNAM-S groups) exhibited significant differences in immune status, CSC, mutation and drug sensitivity. Conclusion: The findings of the study demonstrate the clinical value of RNA modifications, which provide new insights into the individualized treatment for patients with HCC.

| [1] |
Z. Xu, B. Peng, Q. Liang, X. Chen, Y. Cai, S. Zeng, et al., Construction of a ferroptosis-related nine-lncRNA signature for predicting prognosis and immune response in hepatocellular carcinoma, Front. Immunol., 12 (2021), 719175. https://doi.org/10.3389/fimmu.2021.719175 doi: 10.3389/fimmu.2021.719175

|
| [2] |
A. Villanueva, Hepatocellular carcinoma, N. Engl. J. Med., 380 (2019), 1450–1462. https://doi.org/10.1056/NEJMra1713263 doi: 10.1056/NEJMra1713263
|
| [3] |
K. A. McGlynn, J. L. Petrick, H. B. El-Serag, Epidemiology of hepatocellular carcinoma, Hepatology, 73 (2021), 4–13. https://doi.org/10.1002/hep.31288 doi: 10.1002/hep.31288
|
| [4] |
N. Minaei, R. Ramezankhani, A. Tamimi, A. Piryaei, A. Zarrabi, A. R. Aref, et al., Immunotherapeutic approaches in hepatocellular carcinoma: Building blocks of hope in near future, Eur. J. Cell Biol., 102 (2023), 151284. https://doi.org/10.1016/j.ejcb.2022.151284 doi: 10.1016/j.ejcb.2022.151284
|
| [5] |
A. J. Craig, J. von Felden, T. Garcia-Lezana, S. Sarcognato, A. Villanueva, Tumour evolution in hepatocellular carcinoma, Nat. Rev. Gastroenterol. Hepatol., 17 (2020), 139–152. https://doi.org/10.1038/s41575-019-0229-4 doi: 10.1038/s41575-019-0229-4
|
| [6] |
L. K. Chan, Y. M. Tsui, D. W. Ho, I. O. Ng, Cellular heterogeneity and plasticity in liver cancer, Semin. Cancer Biol., 82 (2022), 134–149. https://doi.org/10.1016/j.semcancer.2021.02.015 doi: 10.1016/j.semcancer.2021.02.015
|
| [7] |
I. Barbieri, T. Kouzarides, Role of RNA modifications in cancer, Nat. Rev. Cancer, 20 (2020), 303–322. https://doi.org/10.1038/s41568-020-0253-2 doi: 10.1038/s41568-020-0253-2
|
| [8] |
D. Benak, S. Benakova, L. Plecita-Hlavata, M. Hlavackova, The role of m(6)A and m(6)Am RNA modifications in the pathogenesis of diabetes mellitus, Front. Endocrinol. (Lausanne), 14 (2023), 1223583. https://doi.org/10.3389/fendo.2023.1223583 doi: 10.3389/fendo.2023.1223583
|
| [9] |
S. H. Chung, T. N. Sin, B. Dang, T. Ngo, T. Lo, D. Lent-Schochet, et al., CRISPR-based VEGF suppression using paired guide RNAs for treatment of choroidal neovascularization, Mol. Ther. Nucleic Acids, 28 (2022), 613–622. https://doi.org/10.1016/j.omtn.2022.04.015 doi: 10.1016/j.omtn.2022.04.015
|
| [10] |
S. H. Chung, I. N. Mollhoff, U. Nguyen, A. Nguyen, N. Stucka, E. Tieu, et al., Factors impacting efficacy of AAV-mediated CRISPR-based genome editing for treatment of choroidal neovascularization, Mol. Ther. Methods Clin. Dev., 17 (2020), 409–417. https://doi.org/10.1016/j.omtm.2020.01.006 doi: 10.1016/j.omtm.2020.01.006
|
| [11] |
X. Y. Chen, J. Zhang, J. S. Zhu, The role of m(6)A RNA methylation in human cancer, Mol. Cancer, 18 (2019), 103. https://doi.org/10.1186/s12943-019-1033-z doi: 10.1186/s12943-019-1033-z
|
| [12] |
P. Nombela, B. Miguel-López, S. Blanco, The role of m(6)A, m(5)C and Ψ RNA modifications in cancer: Novel therapeutic opportunities, Mol. Cancer, 20 (2021), 18. https://doi.org/10.1186/s12943-020-01263-w doi: 10.1186/s12943-020-01263-w
|
| [13] |
Q. Zheng, X. Yu, Q. Zhang, Y. He, W. Guo, Genetic characteristics and prognostic implications of m1A regulators in pancreatic cancer, Biosci. Rep., 41 (2021). https://doi.org/10.1042/BSR20210337 doi: 10.1042/BSR20210337
|
| [14] |
Q. Zhang, F. Liu, W. Chen, H. Miao, H. Liang, Z. Liao, et al., The role of RNA m(5)C modification in cancer metastasis, Int. J. Biol. Sci., 17 (2021), 3369–3380. https://doi.org/10.7150/ijbs.61439 doi: 10.7150/ijbs.61439
|
| [15] |
T. Sun, R. Wu, L. Ming, The role of m6A RNA methylation in cancer, Biomed. Pharmacother., 112 (2019), 108613. https://doi.org/10.1016/j.biopha.2019.108613 doi: 10.1016/j.biopha.2019.108613
|
| [16] |
Y. Luo, Y. Yao, P. Wu, X. Zi, N. Sun, J. He, The potential role of N(7)-methylguanosine (m7G) in cancer, J. Hematol. Oncol., 15 (2022), 63. https://doi.org/10.1186/s13045-022-01285-5 doi: 10.1186/s13045-022-01285-5
|
| [17] |
Y. Wang, J. Wang, X. Li, X. Xiong, J. Wang, Z. Zhou, et al., N(1)-methyladenosine methylation in tRNA drives liver tumourigenesis by regulating cholesterol metabolism, Nat. Commun., 12 (2021), 6314. https://doi.org/10.1038/s41467-021-26718-6 doi: 10.1038/s41467-021-26718-6
|
| [18] |
C. Xue, Y. Zhao, G. Li, L. Li, Multi-Omic Analyses of the m(5)C Regulator ALYREF reveal its essential roles in hepatocellular carcinoma, Front. Oncol., 11 (2021), 633415. https://doi.org/10.3389/fonc.2021.633415 doi: 10.3389/fonc.2021.633415
|
| [19] | Y. He, X. Yu, J. Li, Q. Zhang, Q. Zheng, W. Guo, Role of m(5)C-related regulatory genes in the diagnosis and prognosis of hepatocellular carcinoma, Am. J. Transl. Res., 12 (2020), 912–922. |
| [20] |
J. Liu, K. Jiang, METTL3-mediated maturation of miR-589-5p promotes the malignant development of liver cancer, J. Cell. Mol. Med., 26 (2022), 2505–2519. https://doi.org/10.1111/jcmm.16845 doi: 10.1111/jcmm.16845
|
| [21] |
Z. Dai, H. Liu, J. Liao, C. Huang, X. Ren, W. Zhu, et al., N(7)-Methylguanosine tRNA modification enhances oncogenic mRNA translation and promotes intrahepatic cholangiocarcinoma progression, Mol. Cell, 81 (2021), 3339–3355. https://doi.org/10.1016/j.molcel.2021.07.003 doi: 10.1016/j.molcel.2021.07.003
|
| [22] |
Y. Xu, M. Zhang, Q. Zhang, X. Yu, Z. Sun, Y. He, et al., Role of main RNA methylation in hepatocellular carcinoma: N6-Methyladenosine, 5-Methylcytosine, and N1-Methyladenosine, Front. Cell Dev. Biol., 9 (2021), 767668. https://doi.org/10.3389/fcell.2021.767668 doi: 10.3389/fcell.2021.767668
|
| [23] |
C. Tomikawa, 7-Methylguanosine modifications in transfer RNA (tRNA), Int. J. Mol. Sci., 19 (2018). https://doi.org/10.3390/ijms19124080 doi: 10.3390/ijms19124080
|
| [24] |
Y. Zhou, B. Zhou, L. Pache, M. Chang, A. H. Khodabakhshi, O. Tanaseichuk, et al., Metascape provides a biologist-oriented resource for the analysis of systems-level datasets, Nat. Commun., 10 (2019), 1523. https://doi.org/10.1038/s41467-019-09234-6 doi: 10.1038/s41467-019-09234-6
|
| [25] |
M. J. Bywater, R. B. Pearson, G. A. McArthur, R. D. Hannan, Dysregulation of the basal RNA polymerase transcription apparatus in cancer, Nat. Rev. Cancer, 13 (2013), 299–314. https://doi.org/10.1038/nrc3496 doi: 10.1038/nrc3496
|
| [26] |
L. A. Garraway, E. S. Lander, Lessons from the cancer genome, Cell, 153 (2013), 17–37. https://doi.org/10.1016/j.cell.2013.03.002 doi: 10.1016/j.cell.2013.03.002
|
| [27] |
J. A. Joyce, J. W. Pollard, Microenvironmental regulation of metastasis, Nat. Rev. Cancer, 9 (2009), 239–252. https://doi.org/10.1038/nrc2618 doi: 10.1038/nrc2618
|
| [28] |
D. Hanahan, L. M. Coussens, Accessories to the crime: Functions of cells recruited to the tumor microenvironment, Cancer Cell, 21 (2012), 309–322. https://doi.org/10.1016/j.ccr.2012.02.022 doi: 10.1016/j.ccr.2012.02.022
|
| [29] |
T. M. Malta, A. Sokolov, A. J. Gentles, T. Burzykowski, L. Poisson, J. N. Weinstein, et al., Machine learning identifies stemness features associated with oncogenic dedifferentiation, Cell, 173 (2018), 338–354. https://doi.org/10.1016/j.cell.2018.03.034 doi: 10.1016/j.cell.2018.03.034
|
| [30] |
A. Iasonos, D. Schrag, G. V. Raj, K. S. Panageas, How to build and interpret a nomogram for cancer prognosis, J. Clin. Oncol., 26 (2008), 1364–1370. https://doi.org/10.1200/JCO.2007.12.9791 doi: 10.1200/JCO.2007.12.9791
|
| [31] |
Z. Yang, Q. Zi, K. Xu, C. Wang, Q. Chi, Development of a macrophages-related 4-gene signature and nomogram for the overall survival prediction of hepatocellular carcinoma based on WGCNA and LASSO algorithm, Int. Immunopharmacol., 90 (2021), 107238. https://doi.org/10.1016/j.intimp.2020.107238 doi: 10.1016/j.intimp.2020.107238
|
| [32] |
A. Dirican, D. Uncu, M. Sekacheva, M. Artaç, A. Aladashvil, A. Erdogan, et al., A multicentre, multinational study of clinical characteristics and prognosis of hepatocellular carcinoma, East. Mediterr. Health J., 29 (2023), 462–473. https://doi.org/10.26719/emhj.23.087 doi: 10.26719/emhj.23.087
|
| [33] |
C. Xie, X. Ye, L. Zeng, X. Zeng, D. Cao, Serum AKR1B10 as an indicator of unfavorable survival of hepatocellular carcinoma, J. Gastroenterol., (2023). https://doi.org/10.1007/s00535-023-02011-9 doi: 10.1007/s00535-023-02011-9
|
| [34] |
M. Chen, L. Wei, C. T. Law, F. H. Tsang, J. Shen, C. L. Cheng, et al., RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2, Hepatology, 67 (2018), 2254–2270. https://doi.org/10.1002/hep.29683 doi: 10.1002/hep.29683
|
| [35] |
D. Li, K. Li, W. Zhang, K. W. Yang, D. A. Mu, G. J. Jiang, et al., The m6A/m5C/m1A regulated gene signature predicts the prognosis and correlates with the immune status of hepatocellular carcinoma, Front Immunol, 13 (2022), 918140. https://doi.org/10.3389/fimmu.2022.918140 doi: 10.3389/fimmu.2022.918140
|
| [36] |
M. Xiong, K. Zhuang, Y. Luo, Q. Lai, X. Luo, Y. Fang, et al., KIF20A promotes cellular malignant behavior and enhances resistance to chemotherapy in colorectal cancer through regulation of the JAK/STAT3 signaling pathway, Aging, 11 (2019), 11905–11921. https://doi.org/10.18632/aging.102505 doi: 10.18632/aging.102505
|
| [37] |
X. Meng, W. Li, H. Yuan, W. Dong, W. Xiao, X. Zhang, KDELR2-KIF20A axis facilitates bladder cancer growth and metastasis by enhancing Golgi-mediated secretion, Biol. Proced. Online, 24 (2022), 12. https://doi.org/10.1186/s12575-022-00174-y doi: 10.1186/s12575-022-00174-y
|
| [38] |
V. A. Copello, K. L. Burnstein, The kinesin KIF20A promotes progression to castration-resistant prostate cancer through autocrine activation of the androgen receptor, Oncogene, 41 (2022), 2824–2832. https://doi.org/10.1038/s41388-022-02307-9 doi: 10.1038/s41388-022-02307-9
|
| [39] |
C. Wu, X. Qi, Z. Qiu, G. Deng, L. Zhong, Low expression of KIF20A suppresses cell proliferation, promotes chemosensitivity and is associated with better prognosis in HCC, Aging, 13 (2021), 22148–22163. https://doi.org/10.18632/aging.203494 doi: 10.18632/aging.203494
|
| [40] |
Y. Hu, C. Tang, W. Zhu, H. Ye, Y. Lin, R. Wang, et al., Identification of chromosomal instability-associated genes as hepatocellular carcinoma progression-related biomarkers to guide clinical diagnosis, prognosis and therapy, Comput. Biol. Med., 148 (2022), 105896. https://doi.org/10.1016/j.compbiomed.2022.105896 doi: 10.1016/j.compbiomed.2022.105896
|
| [41] |
N. Ouyang, S. Ke, N. Eagleton, Y. Xie, G. Chen, B. Laffins, et al., Pregnane X receptor suppresses proliferation and tumourigenicity of colon cancer cells, Br. J. Cancer, 102 (2010), 1753–1761. https://doi.org/10.1038/sj.bjc.6605677 doi: 10.1038/sj.bjc.6605677
|
| [42] |
Y. Niu, Z. Wang, H. Huang, S. Zhong, W. Cai, Y. Xie, et al., Activated pregnane X receptor inhibits cervical cancer cell proliferation and tumorigenicity by inducing G2/M cell-cycle arrest, Cancer Lett., 347 (2014), 88–97. https://doi.org/10.1016/j.canlet.2014.01.026 doi: 10.1016/j.canlet.2014.01.026
|
| [43] |
X. Niu, T. Wu, G. Li, X. Gu, Y. Tian, H. Cui, Insights into the critical role of the PXR in preventing carcinogenesis and chemotherapeutic drug resistance, Int. J. Biol. Sci., 18 (2022), 742–759. https://doi.org/10.7150/ijbs.68724 doi: 10.7150/ijbs.68724
|
| [44] |
B. D. Khalil, R. Sanchez, T. Rahman, C. Rodriguez-Tirado, S. Moritsch, A. R. Martinez, et al., An NR2F1-specific agonist suppresses metastasis by inducing cancer cell dormancy, J. Exp. Med., 219 (2022). https://doi.org/10.1084/jem.20210836 doi: 10.1084/jem.20210836
|
| [45] |
Y. Liu, P. Zhang, Q. Wu, H. Fang, Y. Wang, Y. Xiao, et al., Long non-coding RNA NR2F1-AS1 induces breast cancer lung metastatic dormancy by regulating NR2F1 and ΔNp63, Nat. Commun., 12 (2021), 5232. https://doi.org/10.1038/s41467-021-25552-0 doi: 10.1038/s41467-021-25552-0
|
| [46] |
D. M. Gilkes, G. L. Semenza, D. Wirtz, Hypoxia and the extracellular matrix: Drivers of tumour metastasis, Nat. Rev. Cancer, 14 (2014), 430–439. https://doi.org/10.1038/nrc3726 doi: 10.1038/nrc3726
|
| [47] |
Z. Wang, G. Fan, H. Zhu, L. Yu, D. She, Y. Wei, et al., PLOD2 high expression associates with immune infiltration and facilitates cancer progression in osteosarcoma, Front. Oncol., 12 (2022), 980390. https://doi.org/10.3389/fonc.2022.980390 doi: 10.3389/fonc.2022.980390
|
| [48] |
Y. Kiyozumi, M. Iwatsuki, J. Kurashige, Y. Ogata, K. Yamashita, Y. Koga, et al., PLOD2 as a potential regulator of peritoneal dissemination in gastric cancer, Int. J. Cancer, 143 (2018), 1202–1211. https://doi.org/10.1002/ijc.31410 doi: 10.1002/ijc.31410
|
| [49] |
T. Noda, H. Yamamoto, I. Takemasa, D. Yamada, M. Uemura, H. Wada, et al., PLOD2 induced under hypoxia is a novel prognostic factor for hepatocellular carcinoma after curative resection, Liver Int., 32 (2012), 110–118. https://doi.org/10.1111/j.1478-3231.2011.02619.x doi: 10.1111/j.1478-3231.2011.02619.x
|
| [50] |
D. C. Hinshaw, L. A. Shevde, The tumor microenvironment innately modulates cancer Progression, Cancer Res., 79 (2019), 4557–4566. https://doi.org/10.1158/0008-5472.CAN-18-3962 doi: 10.1158/0008-5472.CAN-18-3962
|
| [51] |
N. Woller, S. A. Engelskircher, T. Wirth, H. Wedemeyer, Prospects and challenges for T cell-based therapies of HCC, Cells, 10 (2021). https://doi.org/10.3390/cells10071651 doi: 10.3390/cells10071651
|
| [52] |
C. Zheng, L. Zheng, J. K. Yoo, H. Guo, Y. Zhang, X. Guo, et al., Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing, Cell, 169 (2017), 1342–1356. https://doi.org/10.1016/j.cell.2017.05.035 doi: 10.1016/j.cell.2017.05.035
|
| [53] |
Y. Chen, Z. Tian, HBV-induced immune imbalance in the development of HCC, Front. Immunol., 10 (2019), 2048. https://doi.org/10.3389/fimmu.2019.02048 doi: 10.3389/fimmu.2019.02048
|
| [54] |
I. Lurje, L. Hammerich, F. Tacke, Dendritic cell and T cell crosstalk in liver fibrogenesis and hepatocarcinogenesis: implications for prevention and therapy of liver cancer, Int. J. Mol. Sci., 21 (2020). https://doi.org/10.3390/ijms21197378 doi: 10.3390/ijms21197378
|
| [55] |
Z. Tang, T. Zhang, B. Yang, J. Su, Q. Song, SpaCI: deciphering spatial cellular communications through adaptive graph model, Brief. Bioinf., 24 (2023), bbac563. https://doi.org/10.1093/bib/bbac563 doi: 10.1093/bib/bbac563
|
| [56] |
T. Calandra, R. Bucala, Macrophage migration inhibitory factor (mif): A glucocorticoid counter-regulator within the immune system, Crit. Rev. Immunol., 37 (2017), 359–370. https://doi.org/10.1615/CritRevImmunol.v37.i2-6.90 doi: 10.1615/CritRevImmunol.v37.i2-6.90
|
| [57] |
R. K. Meleppat, C. R. Fortenbach, Y. Jian, E. S. Martinez, K. Wagner, B. S. Modjtahedi, et al., In vivo imaging of retinal and choroidal morphology and vascular plexuses of vertebrates using swept-source optical coherence tomography, Transl. Vis. Sci. Technol., 11 (2022), 11. https://doi.org/10.1167/tvst.11.8.11 doi: 10.1167/tvst.11.8.11
|
| [58] |
R. K. Meleppat, K. E. Ronning, S. J. Karlen, K. K. Kothandath, M. E. Burns, E. N. P. Jr, et al., In situ morphologic and spectral characterization of retinal pigment epithelium organelles in mice using multicolor confocal fluorescence imaging, Invest. Ophthalmol. Vis. Sci., 61 (2020), 1. https://doi.org/10.1167/iovs.61.13.1 doi: 10.1167/iovs.61.13.1
|
| [59] |
P. L. Triozzi, E. R. Stirling, Q. Song, B. Westwood, M. Kooshki, M. E. Forbes, et al., Circulating immune bioenergetic, metabolic, and genetic signatures predict melanoma patients' response to anti-pd-1 immune checkpoint blockade, Clin. Cancer Res., 28 (2022), 1192–1202. https://doi.org/10.1158/1078-0432.CCR-21-3114 doi: 10.1158/1078-0432.CCR-21-3114
|
 mbe-20-10-814-Supplementray_Tables.xlsx mbe-20-10-814-Supplementray_Tables.xlsx |
 |
| mbe-20-10-814-Supplementary_Figures.docx |
|










Figures(11)

Yuanqian Yao, Jianlin Lv, Guangyao Wang, Xiaohua Hong. Multi-omics analysis and validation of the tumor microenvironment of hepatocellular carcinoma under RNA modification patterns[J]. Mathematical Biosciences and Engineering, 2023, 20(10): 18318-18344. doi: 10.3934/mbe.2023814







 DownLoad:
DownLoad: