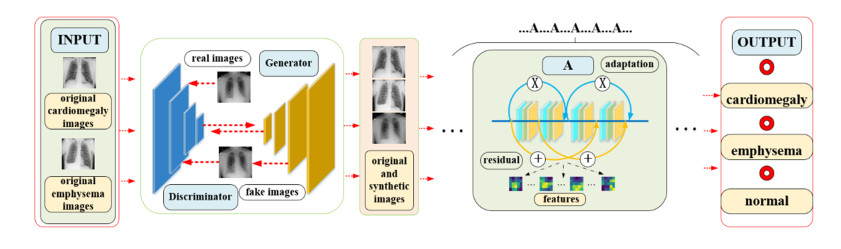
Cardiothoracic diseases are a serious threat to human health and chest X-ray image is a great reference in diagnosis and treatment. At present, it has been a research hot-spot how to recognize chest X-ray image automatically and exactly by the computer vision technology, and many scholars have gotten the excited research achievements. While both emphysema and cardiomegaly often are associated, and the symptom of them are very similar, so the X-ray images discrimination for them led easily to misdiagnosis too. Therefore, some efforts are still expected to develop a higher precision and better performance deep learning model to recognize efficiently the two diseases. In this work, we construct an adaptive cross-transfer residual neural network (ACRnet) to identify emphysema, cardiomegaly and normal. We cross-transfer the information extracted by the residual block and adaptive structure to different levels in ACRnet, and the method avoids the reduction of the adaptive function by residual structure and improves the recognition performance of the model. To evaluate the recognition ability of ACRnet, four neural networks VGG16, InceptionV2, ResNet101 and CliqueNet are used for comparison. The results show that ACRnet has better recognition ability than other networks. In addition, we use the deep convolution generative adversarial network (DCGAN) to expand the original dataset and ACRnet's recognition ability is greatly improved.
Citation: Boyang Wang, Wenyu Zhang. ACRnet: Adaptive Cross-transfer Residual neural network for chest X-ray images discrimination of the cardiothoracic diseases[J]. Mathematical Biosciences and Engineering, 2022, 19(7): 6841-6859. doi: 10.3934/mbe.2022322
Cardiothoracic diseases are a serious threat to human health and chest X-ray image is a great reference in diagnosis and treatment. At present, it has been a research hot-spot how to recognize chest X-ray image automatically and exactly by the computer vision technology, and many scholars have gotten the excited research achievements. While both emphysema and cardiomegaly often are associated, and the symptom of them are very similar, so the X-ray images discrimination for them led easily to misdiagnosis too. Therefore, some efforts are still expected to develop a higher precision and better performance deep learning model to recognize efficiently the two diseases. In this work, we construct an adaptive cross-transfer residual neural network (ACRnet) to identify emphysema, cardiomegaly and normal. We cross-transfer the information extracted by the residual block and adaptive structure to different levels in ACRnet, and the method avoids the reduction of the adaptive function by residual structure and improves the recognition performance of the model. To evaluate the recognition ability of ACRnet, four neural networks VGG16, InceptionV2, ResNet101 and CliqueNet are used for comparison. The results show that ACRnet has better recognition ability than other networks. In addition, we use the deep convolution generative adversarial network (DCGAN) to expand the original dataset and ACRnet's recognition ability is greatly improved.

| [1] |
D. Brenner, J. McLaughlin, R. Hung, Previous lung diseases and lung cancer risk: A systematic review and meta-analysis, PLoS One, 6 (2011). https://doi.org/10.1371/journal.pone.0017479 doi: 10.1371/journal.pone.0017479

|
| [2] |
A. Krizhevsky, I. Sutskever, G. E. Hinton, ImageNet classification with deep convolutional neural networks, Commun. ACM, 60 (2017), 84–90. https://doi.org/10.1371/journal.pone.001747910.1145/3065386 doi: 10.1371/journal.pone.001747910.1145/3065386
|
| [3] |
O. Russakovsky, J. Deng, H. Su, J. Krause, S. Satheesh, S. Ma, et al., ImageNet Large Scale Visual Recognition Challenge, Int. J. Comput. Vision, 115 (2015), 211–252. https://doi.org/10.1007/s11263-015-0816-y doi: 10.1007/s11263-015-0816-y
|
| [4] |
M. Everingham, S. M. A. Eslami, L. Van Gool, C. K. I. Williams, J. Winn, A. Zisserman, The pascal visual object classes challenge: A retrospective, Int. J. Comp. Vision, 111 (2014), 98–136, https://doi.org/10.1007/s11263-014-0733-5 doi: 10.1007/s11263-014-0733-5
|
| [5] | T. Y. Lin, M. Maire, S. Belongie, J. Hays, P. Perona, D. Ramanan, et al., Microsoft COCO: Common objects in context, in Computer Vision – ECCV 2014, Springer, 8693 (2014), 740–755. https://doi.org/10.1007/978-3-319-10602-1_48 |
| [6] |
L. Zhang, P. Yang, H. Feng, Q. Zhao, H. Liu, Using network distance analysis to predict lncRNA-miRNA interactions, Interdiscip. Sci., 13 (2021), 535–545. https://doi.org/10.1007/s12539-021-00458-z doi: 10.1007/s12539-021-00458-z
|
| [7] |
G. Liang, L. Zheng, A transfer learning method with deep residual network for pediatric pneumonia diagnosis, Comput. Methods Programs Biomed., 187 (2020). https://doi.org/10.1016/j.cmpb.2019.06.023 doi: 10.1016/j.cmpb.2019.06.023
|
| [8] | X. Wei, Y. Chen, Z. Zhang, Comparative experiment of convolutional neural network (CNN) models based on pneumonia X-ray images detection, in 2020 2nd International Conference on Machine Learning, Big Data and Business Intelligence (MLBDBI), (2020), 449–454. https://doi.org/10.1109/MLBDBI51377.2020.00095 |
| [9] | L. Račić, T. Popovic, S. Caki, S. Sandi, Pneumonia detection using deep learning based on convolutional neural network, in 2021 25th International Conference on Information Technology, (2021), 1–4. https://doi.org/10.1109/IT51528.2021.9390137 |
| [10] |
A. G. Taylor, C. Mielke, J. Mongan, Automated detection of moderate and large pneumothorax on frontal chest X-rays using deep convolutional neural networks: A retrospective study, PLoS Med., 15 (2018). https://doi.org/10.1371/journal.pmed.1002697 doi: 10.1371/journal.pmed.1002697
|
| [11] | T. K. K. Ho, J. Gwak, O. Prakash, J. I. Song, C. M. Park, Utilizing pretrained deep learning models for automated pulmonary tuberculosis detection using chest radiography, in Intelligent Information and Database Systems, Springer, 11432 (2019), 395–403. https://doi.org/10.1007/978-3-030-14802-7_34 |
| [12] | R. Zhang, M. Sun, S. Wang, K. Chen, Computed Tomography pulmonary nodule detection method based on deep learning, US 10937157B2, L. Infervision Medical Technology, 2021. Available from: https://patentimages.storage.googleapis.com/9c/00/cc/4c302cd759496a/US10937157.pdf. |
| [13] |
C. Tong, B. Liang, Q. Su, M. Yu, J. Hu, A. K. Bashir, et al., Pulmonary nodule classification based on heterogeneous features learning, IEEE J. Sel. Areas Commun., 39 (2021), 574–581. https://doi.org/10.1109/JSAC.2020.3020657 doi: 10.1109/JSAC.2020.3020657
|
| [14] |
L. J. Hyuk, S. H. Young, P. Sunggyun, K. Hyungjin, H. E. Jin, G. J. Mo, et al., Performance of a deep learning algorithm compared with radiologic interpretation for lung cancer detection on chest radiographs in a health screening population, Radiology, 297 (2020), 687–696. https://doi.org/10.1148/radiol.2020201240 doi: 10.1148/radiol.2020201240
|
| [15] |
A. Hosny, C. Parmar, T. P. Coroller, P. Grossmann, R. Zeleznik, A. Kumar, et al., Deep learning for lung cancer prognostication: a retrospective multi-cohort radiomics study, PLoS Med., 15 (2018). https://doi.org/10.1371/journal.pmed.1002711 doi: 10.1371/journal.pmed.1002711
|
| [16] |
M. Masud, N. Sikder, A. A. Nahid, A. K. Bairagi, M. A. Alzain, A machine learning approach to diagnosing lung and colon cancer using a deep learningbased classification framework, Sensors (Basel), 21 (2021), 1–21. https://doi.org/10.3390/s21030748 doi: 10.3390/s21030748
|
| [17] |
S. Roy, W. Menapace, S. Oei, B. Luijten, E. Fini, C. Saltori, et al., Deep learning for classification and localization of COVID-19 markers in point-of-care lung ultrasound, IEEE Trans. Med. Imaging, 39 (2020), 2676–2687. https://doi.org/10.1109/TMI.2020.2994459 doi: 10.1109/TMI.2020.2994459
|
| [18] |
H. T. Qing, K. Mohammad, M. Mokhtar, P. GholamReza, T. Karim, T. A. Rashid, Real‑time COVID-19 diagnosis from X-Ray images using deep CNN and extreme learning machines stabilized by chimp optimization algorithm, Biomed. Signal Process. Control, 68 (2021). https://doi.org/10.1016/j.bspc.2021.102764 doi: 10.1016/j.bspc.2021.102764
|
| [19] |
M. A. Khan, S. Kadry, Y. D. Zhang, T. Akram, M. Sharif, A. Rehman, et al., Prediction of COVID-19 - pneumonia based on selected deep features and one class kernel extreme learning machine, Comput. Electr. Eng., 90 (2021). https://doi.org/10.1016/j.compeleceng.2020.106960 doi: 10.1016/j.compeleceng.2020.106960
|
| [20] | Y. Qasim, B. Ahmed, T. Alhadad, H. A. Sameai, O. Ali, The impact of data augmentation on accuracy of COVID-19 detection based on X-ray images, in Innovative Systems for Intelligent Health Informatics, Lecture Notes on Data Engineering and Communications Technologies, Springer, 72 (2021), 1041–1049. https://doi.org/10.1007/978-3-030-70713-2_93 |
| [21] |
M. Loey, F. Smarandache, N. E. M. Khalifa, Within the lack of chest COVID-19 X-ray dataset: A novel detection model based on GAN and deep transfer learning, Symmetry, 12 (2020). https://doi.org/10.3390/sym12040651 doi: 10.3390/sym12040651
|
| [22] |
S. Y. Lu, D. Wu, Z. Zhang, S. H. Wang, An explainable framework for diagnosis of COVID-19 pneumonia via transfer learning and discriminant correlation analysis, ACM Trans. Multimedia Comput. Commun. Appl., 17 (2021), 1–16. https://doi.org/10.1145/3449785 doi: 10.1145/3449785
|
| [23] |
S. Y. Lu, Z. Q. Zhu, J. M. Gorriz, S. H. Wang, Y. D. Zhang, NAGNN: Classification of COVID-19 based on neighboring aware representation from deep graph neural network, Int. J. Intell. Syst., 37 (2021), 1572–1598. https://doi.org/10.1002/int.22686 doi: 10.1002/int.22686
|
| [24] |
L. T. Duong, N. H. Le, T. B. Tran, V. M. Ngo, P. T. Nguyen, Detection of tuberculosis from chest X-ray images: boosting the performance with vision transformer and transfer learning, Expert Syst. Appl., 184 (2021), 115519. https://doi.org/10.1016/j.eswa.2021.115519 doi: 10.1016/j.eswa.2021.115519
|
| [25] | J. R. F. Junior, D. A. Cardona, R. A. Moreno, M. F. S. Rebelo, J. E. Krieger, M. A. Gutierrez, A general fully automated deep-learning method to detect cardiomegaly in chest x-rays, in Progress in Biomedical Optics and Imaging 2021: Computer-Aided Diagnosis, 2021. https://doi.org/10.1117/12.2581980 |
| [26] |
Y. Wu, S. Qi, Y. Sun, S. Xia, Y. Yao, W. Qian, et al., A vision transformer for emphysema classification using CT images, Phys. Med. Biol., 66 (2021), 245016. https://doi.org/10.1088/1361-6560/ac3dc8 doi: 10.1088/1361-6560/ac3dc8
|
| [27] |
P. Rajpurkar, J. Irvin, R. L. Ball, K. Zhu, B. Yang, H. Mehta, et al., Deep learning for chest radiograph diagnosis: A retrospective comparison of the CheXNeXt algorithm to practicing radiologists, PLoS Med., 15 (2018). https://doi.org/10.1371/journal.pmed.1002686 doi: 10.1371/journal.pmed.1002686
|
| [28] | A. I. A. Rivero, N. Papadakis, R. Li, P. Sellars, Q. Fan, R. T. Tan, et al., GraphX NET-chest X-Ray classification under extreme minimal supervision, in Medical Image Computing and Computer Assisted Intervention–MICCAI 2019-22nd International Conference, 2019. https://doi.org/10.1007/978-3-030-32226-7_56 |
| [29] | X. Wang, Y. Peng, L. Lu, Z. Lu, R. M. Summers, TieNet: Text-image embedding network for common thorax disease classification and reporting in chest X-rays, in Proceedings of the IEEE Computer Society Conference on Computer Vision and Pattern Recognition, (2018), 9049–9058. https://doi.org/10.1109/CVPR.2018.00943 |
| [30] |
J. Zhao, M. Li, W. Shi, Y. Miao, Z. Jiang, B. Ji, A deep learning method for classification of chest X-ray images, J. Phys. Conf. Ser., 1848 (2021). https://doi.org/10.1088/1742-6596/1848/1/012030 doi: 10.1088/1742-6596/1848/1/012030
|
| [31] |
T. K. K. Ho, J. Gwak, Utilizing knowledge distillation in deep learning for classification of chest X-ray abnormalities, IEEE Access, 8 (2020), 160749–160761. https://doi.org/10.1109/ACCESS.2020.3020802 doi: 10.1109/ACCESS.2020.3020802
|
| [32] | Y. Xiao, M. Lu, Z. Fu, Covered face recognition based on deep convolution generative adversarial networks, in Lecture Notes in Computer Science, (2020), 133–141. https://doi.org/10.1007/978-3-030-57884-8_12 |
| [33] | K. Simonyan, A. Zisserman, Very deep convolutional networks for large-scale image recognition, in 3rd International Conference on Learning Representations, ICLR 2015 - Conference Track Proceedings, 2015. https://doi.org/10.1109/acpr.2015.7486599 |
| [34] | C. Szegedy, W. Liu, Y. Jia, P. Sermanet, S. Reed, D. Anguelov, et al, Going deeper with convolutions, in Proceedings of the IEEE Computer Society Conference on Computer Vision and Pattern Recognition, (2015), 1–9. https://doi.org/10.1109/cvpr.2015.7298594 |
| [35] | S. Ioffe, C. Szegedy, Batch normalization: Accelerating deep network training by reducing internal covariate shift, in 32nd International Conference on Machine Learning, (2015), 448–456. Available from: http://proceedings.mlr.press/v37/ioffe15.pdf. |
| [36] | K. He, X. Zhang, S. Ren, J. Sun, Deep residual learning for image recognition, in Proceedings of the IEEE Computer Society Conference on Computer Vision and Pattern Recognition, (2016), 770–778. https://doi.org/10.1109/cvpr.2016.90 |
| [37] | Y. Yang, Z. Zhong, T. Shen, Z. Lin, Convolutional neural networks with alternately updated clique, in Proceedings of the IEEE Computer Society Conference on Computer Vision and Pattern Recognition, (2018), 2413–2422. https://doi.org/10.1109/CVPR.2018.00256 |
| [38] |
J. Hu, L. Shen, S. Albanie, G. Sun, E. Wu, Squeeze-and-excitation networks, IEEE Tra-ns. Pattern Anal. Mach. Intell., 42 (2020), 2011–2023. https://doi.org/10.1109/TPAMI.2019.2913372 doi: 10.1109/TPAMI.2019.2913372
|
| [39] | G. E. Hinton, N. Srivastava, A. Krizhevsky, I. Sutskever, R. R. Salakhutdinov, Improving neural networks by preventing co-adaptation of feature detectors, preprint, arXiv: 1207.0580v1. |
| [40] | T. Salimans, I. Goodfellow, W. Zaremba, V. Cheung, A. Radford, X. Chen, Improved techniques for training GANs, Adv. Neural Inf. Process. Syst., (2016), 2234–2242. Available from: https://proceedings.neurips.cc/paper/2016/file/8a3363abe792db2d8761d6403605aeb7-Paper.pdf. |
| [41] | M. Heusel, H. Ramsauer, T. Unterthiner, B. Nessler, S. Hochreiter, GANs trained by a two time-scale update rule converge to a local Nash equilibrium, Adv. Neural Inf. Pro-cess. Syst., (2017), 6627–6638. Available from: https://proceedings.neurips.cc/paper/2017/file/8a1d694707eb0fefe65871369074926d-Paper.pdf. |
| [42] |
F. F. Li, J. Deng, K. Li, ImageNet: Constructing a large-scale image database, J. Vision, 9 (2009). https://doi.org/10.1167/9.8.1037 doi: 10.1167/9.8.1037
|
| [43] | C. Szegedy, V. Vanhoucke, S. Ioffe, J. Shlens, Z. Wojna, Rethinking the inception architecture for computer vision, in Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition (CVPR), (2016), 2818–2826. https://doi.org/10.1109/cvpr.2016.308 |
| [44] |
N. L. Ramo, K. L. Troyer, C. M. Puttlitz, Comparing predictive accuracy and computational costs for viscoelastic modeling of spinal cord tissues, J. Biomech. Eng., 141 (2019). https://doi.org/10.1115/1.4043033 doi: 10.1115/1.4043033
|
| [45] | D. M. Powers, Evaluation: From precision, recall and f-measure to ROC, informedness, markedness and correlation, J. Mach. Learn. Technol., 2 (2011), 2229–3981. Available from: http://hdl.handle.net/2328/27165. |
| [46] |
T. Fawcett, An introduction to ROC analysis, Pattern Recognit. Lett., 27 (2006), 861–874. https://doi.org/10.1016/j.patrec.2005.10.010 doi: 10.1016/j.patrec.2005.10.010
|
| [47] | C. X. Ling, J. Huang, H. Zhang, AUC: A better measure than accuracy in comparing learning algorithms, in Advances in Artificial Intelligence, 16th Conference of the Canadian Society for Computational Studies of Intelligence, AI 2003, Halifax, Canada, 2003. https://doi.org/10.1007/3-540-44886-1_25 |
| [48] | G. Zeng, On the confusion matrix in credit scoring and its analytical properties, in Communications in Statistics - Theory and Methods, 49 (2020), 2080–2093. https://doi.org/10.1080/03610926.2019.1568485 |





Figures(6) / Tables(7)

Boyang Wang, Wenyu Zhang. ACRnet: Adaptive Cross-transfer Residual neural network for chest X-ray images discrimination of the cardiothoracic diseases[J]. Mathematical Biosciences and Engineering, 2022, 19(7): 6841-6859. doi: 10.3934/mbe.2022322







 DownLoad:
DownLoad: