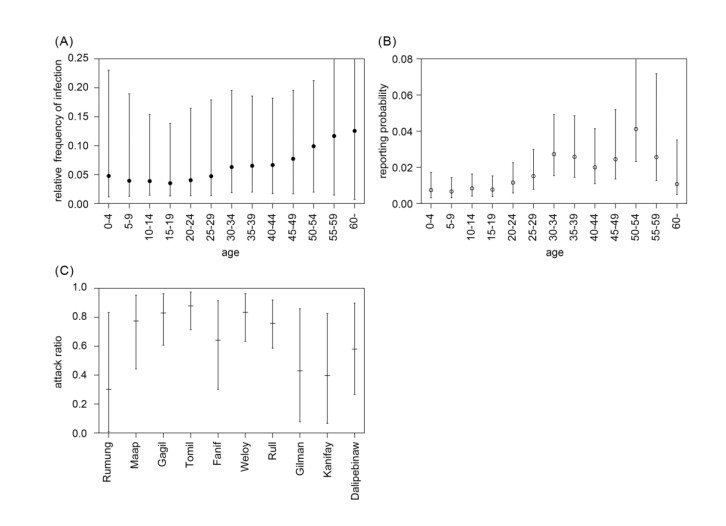
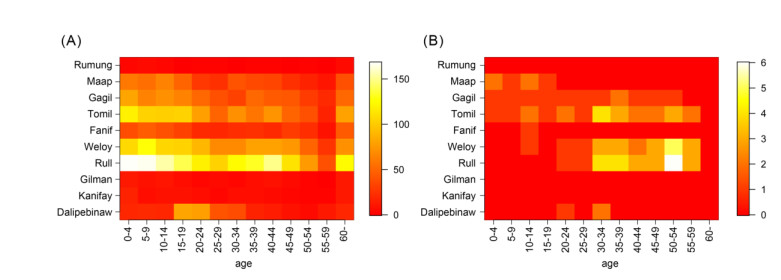
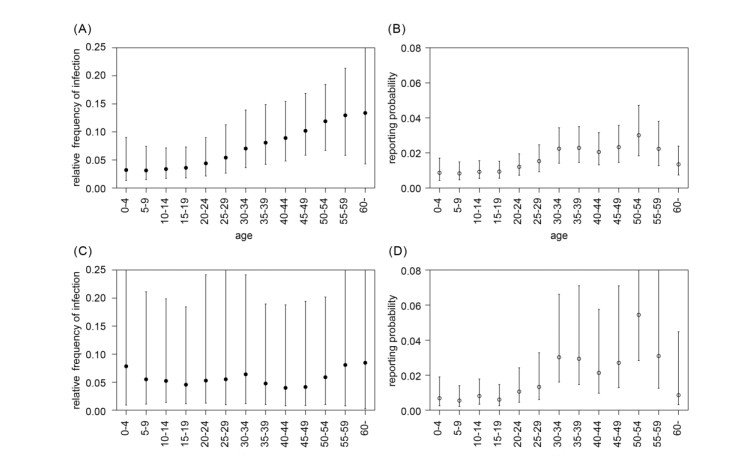
Intensive surveillance of Zika virus infection conducted on Yap Island has provided crucial information on the epidemiological characteristics of the virus, but the rate of infection and medical attendance stratified by age and geographical location of the epidemic have yet to be fully clarified. In the present study, we reanalyzed surveillance data reported in a previous study. Likelihood-based Bayesian inference was used to gauge the age and geographically dependent force of infection and age-dependent reporting rate, with unobservable variables imputed by the data augmentation method. The inferred age-dependent component of the force of infection was suggested to be up to 3-4 times higher among older adults than among children. The age-dependent reporting rate ranged from 0.7% (5-9 years old) to 3.3% (50-54 years old). The proportion of serologically confirmed cases among total probable or confirmed cases was estimated to be 44.9%. The cumulative incidence of infection varied by municipality: Median values were over 80% in multiple locations (Gagil, Tomil, and Weloy), but relatively low values (below 50%) were derived in other locations. However, the possibility of a comparably high incidence of infection was not excluded even in municipalities with the lowest estimates. The results suggested a high degree of heterogeneity in the Yap epidemic. The force of infection and reporting rate were higher among older age groups, and this discrepancy implied that the demographic patterns were remarkably different between all infected and medically attended individuals. A higher reporting rate may have reflected more severe clinical presentation among adults. The symptomatic ratio in dengue cases is known to correlate with age, and our findings presumably indicate a similar tendency in Zika virus disease.
Citation: A. Endo, H. Nishiura. Age and geographic dependence of Zika virus infection during the outbreak on Yap island, 2007[J]. Mathematical Biosciences and Engineering, 2020, 17(4): 4115-4126. doi: 10.3934/mbe.2020228
Intensive surveillance of Zika virus infection conducted on Yap Island has provided crucial information on the epidemiological characteristics of the virus, but the rate of infection and medical attendance stratified by age and geographical location of the epidemic have yet to be fully clarified. In the present study, we reanalyzed surveillance data reported in a previous study. Likelihood-based Bayesian inference was used to gauge the age and geographically dependent force of infection and age-dependent reporting rate, with unobservable variables imputed by the data augmentation method. The inferred age-dependent component of the force of infection was suggested to be up to 3-4 times higher among older adults than among children. The age-dependent reporting rate ranged from 0.7% (5-9 years old) to 3.3% (50-54 years old). The proportion of serologically confirmed cases among total probable or confirmed cases was estimated to be 44.9%. The cumulative incidence of infection varied by municipality: Median values were over 80% in multiple locations (Gagil, Tomil, and Weloy), but relatively low values (below 50%) were derived in other locations. However, the possibility of a comparably high incidence of infection was not excluded even in municipalities with the lowest estimates. The results suggested a high degree of heterogeneity in the Yap epidemic. The force of infection and reporting rate were higher among older age groups, and this discrepancy implied that the demographic patterns were remarkably different between all infected and medically attended individuals. A higher reporting rate may have reflected more severe clinical presentation among adults. The symptomatic ratio in dengue cases is known to correlate with age, and our findings presumably indicate a similar tendency in Zika virus disease.

| [1] |
G. W. A. Dick, S. F. Kitchen, A. J. Haddow, Zika virus (Ⅰ). Isolations and serological specificity, Trans. R. Soc. Trop. Med. Hyg., 46 (1952), 509-520. doi: 10.1016/0035-9203(52)90042-4

|
| [2] |
M. R. Duffy, T. H. Chen, W. T. Hancock, A. M. Powers, J. L. Kool, R. S. Lanciotti, et al., Zika virus outbreak on Yap Island, Federated States of Micronesia, N. Engl. J. Med., 360 (2009), 2536-2543. doi: 10.1056/NEJMoa0805715
|
| [3] | V. M. Cao-Lormeau, C. Roche, A. Teissier, E. Robin, A. L. Berry, H. P. Mallet, Zika virus, French Polynesia, South Pacific, 2013, Emerg. Infect. Dis., 20 (2014), 1085-1086. |
| [4] |
V. M. Cao-Lormeau, D. Musso, 2014. Emerging arboviruses in the Pacific. Lancet., 384 (2014), 1571-1572. doi: 10.1016/S0140-6736(14)61977-2
|
| [5] |
Roth, A. Mercier, C. Lepers, D. Hoy, S. Duituturaga, E. Benyon, et al., Concurrent outbreaks of dengue, chikungunya and Zika virus infections—an unprecedented epidemic wave of mosquito-borne viruses in the Pacific 2012-2014, Euro. Surveill., 19 (2014), 20929. doi: 10.2807/1560-7917.ES2014.19.41.20929
|
| [6] |
G. S. Campos, A. C. Bandeira, S. I. Sardi, Zika virus outbreak, Bahia, Brazil, Emerg. Infect. Dis., 21 (2015), 1885-1886. doi: 10.3201/eid2110.150847
|
| [7] |
M. K. Kindhauser, T. Allen, V. Frank, R. S. Santhana, C. Dye, Zika: the origin and spread of a mosquito-borne virus, Bull. World Health Organ., 94 (2016), 675-686. doi: 10.2471/BLT.16.171082
|
| [8] |
C. Zanluca, V. C. A. de Melo, A. L. P. Mosimann, G. I. V. dos Santos, C. N. D. dos Santos, K. Luz, First report of autochthonous transmission of Zika virus in Brazil, Mem. Inst. Instituto. Oswaldo. Cruz., 110 (2015), 569-572. doi: 10.1590/0074-02760150192
|
| [9] | Pan American Health Organization/World Health Organization, 2017. Zika Epidemiological Update, 26 January 2017. PAHO/WHO, Washington, D.C. |
| [10] |
P. Brasil, J. P. Jr Pereira, M. E. Moreira, R. M. Ribeiro Nogueira, L. Damasceno, M. Wakimoto, et al., Zika virus infection in pregnant women in Rio de Janeiro - preliminary report, N. Engl. J. Med., 375 (2016), 2321-2334. doi: 10.1056/NEJMoa1602412
|
| [11] |
S. Cauchemez, M. Besnard, P. Bompard, T. Dub, P. Guillemette-Artur, D. Eyrolle-Guignot, et al., Association between Zika virus and microcephaly in French Polynesia, 2013-15: a retrospective study, Lancet., 387 (2016), 2125-2132. doi: 10.1016/S0140-6736(16)00651-6
|
| [12] |
W. K. de Oliveira, J. Cortez-Escalante, W. T. G. H. De Oliveira, G. M. I. D. Carmo, C. M. P. Henriques, G. E. Coelho, et al., Increase in reported prevalence of microcephaly in infants born to women living in areas with confirmed zika virus transmission during the first trimester of pregnancy - Brazil, 2015, Morb. Mortal. Wkly Rep., 65 (2016), 242-247. doi: 10.15585/mmwr.mm6509e2
|
| [13] |
F. Krauer, M. Riesen, L. Reveiz, O. T. Oladapo, R. Martínez-Vega, T. V. Porgo, et al., Zika virus infection as a cause of congenital brain abnormalities and Guillain-Barré syndrome: systematic review, PLoS Med., 14 (2017), e1002203. doi: 10.1371/journal.pmed.1002203
|
| [14] |
J. Mlakar, M. Korva, N. Tul, M. Popovic, M. Poljsak-Prijatelj, J. Mraz, et al., Zika virus associated with microcephaly, N. Engl. J. Med., 374 (2016), 951-958. doi: 10.1056/NEJMoa1600651
|
| [15] |
V. M. Cao-Lormeau, A. Blake, S. Mons, S. Lastere, C. Roche, J. Vanhomwegen, et al., Guillain-Barré syndrome outbreak associated with Zika virus infection in French Polynesia: a case-control study, Lancet., 387 (2016), 1531-1539. doi: 10.1016/S0140-6736(16)00562-6
|
| [16] | E. Oehler, L. Watrin, P. Larre, I. Leparc-Goffart, S. Lastere, F. Valour, et al., Zika virus infection complicated by Guillain-Barré syndrome—case report, French Polynesia, December 2013, Euro Surveill., 19 (2014), 20720. |
| [17] |
P. K. Mitchell, L. Mier-Y-Teran-Romero, B. J. Biggerstaff, M. J. Delorey, M. Aubry, V. M. Cao-Lormeau, et al., Reassessing serosurvey-based estimates of the symptomatic proportion of Zika virus infections, Am J Epidemiol., 188 (2019), 206-213. doi: 10.1093/aje/kwy189
|
| [18] | C. Flamand, C. Fritzell, S. Matheus, M. Dueymes, G. Carles, A. Favre, et al., The proportion of asymptomatic infections and spectrum of disease among pregnant women infected by Zika virus: systematic monitoring in French Guiana, 2016, Euro. Surveill., 22 (2017). |
| [19] |
L. Subissi, E. Daudens-Vaysse, S. Cassadou, M. Ledrans, P. Bompard, J. Gustave, et al., Revising rates of asymptomatic Zika virus infection based on sentinel surveillance data from French Overseas Territories, Int. J. Infect. Dis., 65 (2017), 116-118. doi: 10.1016/j.ijid.2017.10.009
|
| [20] | S. Funk, A. J. Kucharski, A. Camacho, R. M. Eggo, L. Yakob, L. M. Murray, et al., Comparative analysis of dengue and Zika outbreaks reveals differences by setting and virus, PLoS Negl. Trop. Dis., 10 (2017), e0005173. |
| [21] |
J. Kucharski, S. Funk, R. M. Eggo, H. P. Mallet, W. J. Edmunds, E. J. Nilles, Transmission dynamics of zika virus in island populations: a modelling analysis of the 2013-14 French Polynesia outbreak, PLoS Negl. Trop. Dis., 10 (2016), e0004726. doi: 10.1371/journal.pntd.0004726
|
| [22] | Micronesia (Federated States), 2002. 2000 FSM Census of Population and Housing. Yap Branch Statistics Office, Division of Statistics, Department of Economic Affairs, Federated States of Micronesia, Colonia, Yap. |
| [23] | W. R. Gilks, S. Richardson, D. J. Spiegelhalter, Markov Chain Monte Carlo in Practice. Chapman and Hall, London, Markov Chain Monte Carlo in practice. Chapman and Hall, London. 1996. |
| [24] |
U. Haque, J. D. Ball, W. Zhang, M. M. Khan, C. J. A. Treviño, 2016. Clinical and spatial features of Zika virus in Mexico, Acta Trop., 162 (2016), 5-10. doi: 10.1016/j.actatropica.2016.06.010
|
| [25] |
J. R. Egger, P. G. Coleman, Age and clinical dengue illness, Emerg. Infect. Dis., 13 (2007), 924-925. doi: 10.3201/eid1306.070008
|
| [26] |
Gordon, G. Kuan, J. C. Mercado, L. Gresh, W. Avilés, A. Balmaseda, et al., The Nicaraguan pediatric dengue cohort study: incidence of inapparent and symptomatic dengue virus infections, 2004-2010, PLoS Negl. Trop. Dis., 7 (2013), e2462. doi: 10.1371/journal.pntd.0002462
|
| [27] |
K. T. Thai, H. Nishiura, P. L. Hoang, N. T. Tran, G. T. Phan, H. Q. Le, et al., Age-specificity of clinical dengue during primary and secondary infections, PLoS Negl. Trop. Dis., 5 (2011), e1180. doi: 10.1371/journal.pntd.0001180
|
| [28] |
Wilder-Smith, W. Foo, A. Earnest, S. Sremulanathan, N. I. Paton, Seroepidemiology of dengue in the adult population of Singapore, Trop. Med. Int. Health., 9 (2004), 305-308. doi: 10.1046/j.1365-3156.2003.01177.x
|
| [29] |
D. A. Cummings, S. Iamsirithaworn, J. T. Lessler, A. McDermott, R. Prasanthong, A. Nisalak, et al., The impact of the demographic transition on dengue in Thailand: insights from a statistical analysis and mathematical modeling, PLoS Med., 6 (2009), e1000139. doi: 10.1371/journal.pmed.1000139
|
| [30] |
Rodríguez-Barraquer, R. Buathong, S. Iamsirithaworn, A. Nisalak, J. Lessler, R. G. Jarman, et al., Revisiting Rayong: shifting seroprofiles of dengue in Thailand and their implications for transmission and control, Am. J. Epidemiol., 179 (2014), 353-360. doi: 10.1093/aje/kwt256
|
| [31] |
H. Muench, Derivation of Rates from Summation Data by the Catalytic Curve, J. Am. Stat. Assoc., 29 (1934), 25-38. doi: 10.1080/01621459.1934.10502684
|
| [32] | H. Muench, Catalytic Models in Epidemiology, 1963. Harvard University Press: Boston (Reprinted in 2013; doi: 10.4159/harvard.9780674428928). |
| [33] |
N. Hens, M. Aerts, C. Faes, Z. Shkedy, O. Lejeune, P. Van Damme, et al., Seventy-five years of estimating the force of infection from current status data, Epidemiol. Infect., 138 (2010), 802-812. doi: 10.1017/S0950268809990781
|
| [34] | W. O. Kermack, A. G. McKendrick, Contributions to the mathematical theory of epidemics-I, Bull. Math. Biol., 53 (1991), 89-118. |
| [35] | K. Lugnér, S. D. Mylius, J. Wallinga, Dynamic versus static models in cost-effectiveness analyses of anti-viral drug therapy to mitigate an influenza pandemic, Health Econ., 19 (2009), 518-531. |
| [36] | Federated States of Micronesia (FSM), Division of Statistics, Summary Analysis of Key Indicators: from the FSM 2010 Census of Population and Housing, Palikir, Pohnpei, 2012. Available from: http://prism.spc.int/images/census_reports/FSM_2010_Census_Indicators_Final.pdf. |
Figures(3) / Tables(2)

A. Endo, H. Nishiura. Age and geographic dependence of Zika virus infection during the outbreak on Yap island, 2007[J]. Mathematical Biosciences and Engineering, 2020, 17(4): 4115-4126. doi: 10.3934/mbe.2020228







 DownLoad:
DownLoad: