Citation: Atefeh Afsar, Filipe Martins, Bruno M. P. M. Oliveira, Alberto A. Pinto. A fit of CD4+ T cell immune response to an infection by lymphocytic choriomeningitis virus[J]. Mathematical Biosciences and Engineering, 2019, 16(6): 7009-7021. doi: 10.3934/mbe.2019352

| [1] | J. Banchereau, F. Briere, C. Caux, et al., Immunobiology of dendritic cells, Annu. Rev. Immunol., 18 (2000), 767–811. |
| [2] | J. Zhu and W. E. Paul, CD4 T cells: fates, functions, and faults, Blood, 112 (2008), 1557–1569. |
| [3] | N. J. Burroughs, B. M. P. M. Oliveira and A. A. Pinto, Regulatory T cell adjustment of quorum growth thresholds and the control of local immune responses, J. Theor. Biol., 241 (2006), 134–141. |
| [4] | A. L. Cava, Tregs are regulated by cytokines: Implications for autoimmunity, Autoimmun. Rev., 8 (2008), 83–87. |
| [5] | B.-I. Moon, T. H. Kim and J.-Y. Seoh, Functional modulation of regulatory T cells by IL-2, PLoS One, 10 (2015), 1–13. |
| [6] | E. M. Shevach, R. S. McHugh, C. A. Piccirillo, et al., Control of T-cell activation by CD4+ CD25+ suppressor T cells, Immunol. Rev., 182 (2001), 58–67. |
| [7] | A. M. Thornton and E. M. Shevach, CD4+ CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukine 2 production, The Journal of Experimental Medicine, 188 (1998), 287–296. |
| [8] | L. A. Turka and P. T. Walsh, IL-2 signaling and CD4+ CD25+ Foxp3+ regulatory T cells, Front. Biosci., 13 (2008), 1440–1446. |
| [9] | S. Sakaguchi, Naturally arising CD4+ regulatory T cells for immunological self-tolerance and negative control of immune responses, Annu. Rev. Immunol., 22 (2004), 531–562. |
| [10] | J. Zhu, H. Yamane and W. E. Paul, Differentiation of Effector CD4 T Cell Populations, Annu. Rev. Immunol., 28 (2010), 445–489. |
| [11] | D. Homann, L. Teyton and M. B. Oldstone, Differential regulation of antiviral T-cell immunity results in stable CD8+ but declining CD4+ T-cell memory, Nat. Med., 7 (2001), 913–919. |
| [12] | R. E. Callard and A. J. Yates, Immunology and mathematics: crossing the divide, Immunology, 115 (2005), 21–33. |
| [13] | G. Lythe and C. Molina-París, Some deterministic and stochastic mathematical models of naïve T-cell homeostasis, Immunol. Rev., 285 (2018), 206–217. |
| [14] | R. J. de Boer and P. Hogeweg, Immunological discrimination between self and non-self by precur-sor depletion and memory accumulation, J. Theor. Biol., 124 (1987), 343–369. |
| [15] | A. Pinto, N. Burroughs, F. Ferreira, et al., Dynamics of immunological models, Acta Biotheor., 58 (2010), 391–404. |
| [16] | K. Blyuss and L. Nicholson, The role of tunable activation thresholds in the dynamics of autoim-munity, J. Theor. Biol., 308 (2012), 45–55. |
| [17] | S. Khailaie, F. Bahrami, M. Janahmadi, et al., A mathematical model of immune activation with a unified self-nonself concept, Front. Immunol., 4 (2013), 474. |
| [18] | K. León, A. Lage, and J. Carneiro, Tolerance and immunity in a mathematical model of T-cell mediated suppression, J. Theor. Biol., 225 (2003), 107–126. |
| [19] | C. Bianca and L. Brézin, Modeling the antigen recognition by B-cell and T-cell receptors through thermostatted kinetic theory methods, Int. J. Biomath., 10 (2017), 1750072. |
| [20] | R. F. Alvarez, J. A. Barbuto and R. Venegeroles, A nonlinear mathematical model of cell-mediated immune response for tumor phenotypic heterogeneity, J. Theor. Biol., 471 (2019), 42–50. |
| [21] | N. Burroughs, M. Ferreira, B. Oliveira, et al., Autoimmunity arising from bystander proliferation of T cells in an immune response model, Math. Comput. Modell., 53 (2011), 1389–1393. |
| [22] | B. M. P. M. Oliveira, R. Trinchet, M. V. Otero-Espinar, et al., Modelling the suppression of au-toimmunity after pathogen infection, Math. Methods Appl. Sci., 41 (2018), 8565–8570. |
| [23] | B. M. P. M. Oliveira, I. P. Figueiredo, N. J. Burroughs, et al., Approximate equilibria for a T cell and Treg model, Appl. Math. Inform. Sci., 9 (2015), 2221–2231. |
| [24] | N. Burroughs, B. Oliveira, A. Pinto, et al., Immune response dynamics, Math. Comput. Modell., 53 (2011), 1410–1419. |
| [25] | N. Burroughs, B. Oliveira, A. Pinto, et al., Sensibility of the quorum growth thresholds controlling local immune responses, Math. Comput. Modell., 47 (2008), 714–725. |
| [26] | N. Burroughs, M. Ferreira, B. Oliveira, et al., A transcritical bifurcation in an immune response model, J. Differ. Eq. Appl., 17 (2011), 1101–1106. |
| [27] | B. M. P. M. Oliveira, A. Yusuf, I. P. Figueiredo, et al., The effect of a linear tuning between the antigenic stimulations of T cells and Tregs. In Preparation. |
| [28] | N. J. Burroughs, M. Ferreira, J. Martins, et al., Dynamics and biological thresholds, in Dynam-ics, Games and Science I, DYNA 2008, in Honor of Maur´ ıcio Peixoto and David Rand (editors A. A. Pinto, D. A. Rand, and M. M. Peixoto), volume 1 of Springer Proceedings in Mathematics, Springer-Verlag Berlin Heidelberg (2011), pp. 183–191. |
| [29] | R. J. de Boer, D. Homann and A. S. Perelson, Different Dynamics of CD4+ and CD8+ T Cell Responses During and After Acute Lymphocytic Choriomeningitis Virus Infection, J. Immunol., 171 (2003), 3928–3935. |
| [30] | R. J. de Boer and A. S. Perelson, Quantifying T lymphocyte turnover, J. Theor. Biol., 327 (2013), 45–87. |
| [31] | R. E. Callard, J. Stark and A. J. Yates, Fratricide: a mechanism for T memory-cell homeostasis, Trends Immunol., 24 (2003), 370–375. |
| [32] | S. Nagata, Fas ligand-induced apoptosis, Annu. Rev. Genet., 33 (1999), 29–55. |
| [33] | P. M. Anderson and M. A. Sorenson, Effects of route and formulation on clinical pharmacokinetics of interleukine-2, Clin. Pharmacokinet., 27 (1994), 19–31. |
| [34] | A. R. Mclean and C. A. Michie, In vivo estimates of division and death rates of human t lympho-cytes., Proceedings of the National Academy of Sciences, 92 (1995), 3707–3711. |
| [35] | C. Michie, A. McLean, C. Alcock, et al., Life-span of human lymphocyte subsets defined by CD45 isoforms, Nature, 360 (1992), 264–265. |
| [36] | D. Moskophidis, M. Battegay, M. Vandenbroek, et al., Role of virus and host variables in virus persistence or immunopathological disease caused by a non-cytolytic virus, J. Gen. Virol., 76 (1995), 381–391. |
| [37] | H. Veiga-Fernandes, U. Walter, C. Bourgeois, et al., Response of naïve and memory CD8+ T cells to antigen stimulation in vivo, Nat. Immunol., 1 (2000), 47–53. 38. G. A. Seber and C. J. Wild, Nonlinear Regression, Wiley and Sons (2003). |
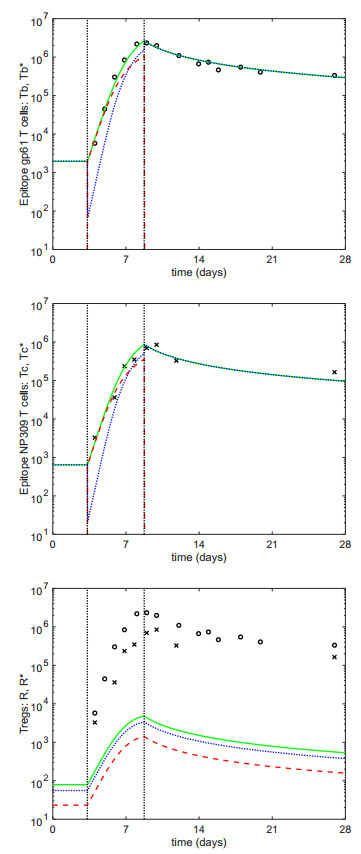
Figures(1) / Tables(2)

Atefeh Afsar, Filipe Martins, Bruno M. P. M. Oliveira, Alberto A. Pinto. A fit of CD4+ T cell immune response to an infection by lymphocytic choriomeningitis virus[J]. Mathematical Biosciences and Engineering, 2019, 16(6): 7009-7021. doi: 10.3934/mbe.2019352







 DownLoad:
DownLoad: