Citation: Eric Ke Wang, Fan Wang, Ruipei Sun, Xi Liu. A new privacy attack network for remote sensing images classification with small training samples[J]. Mathematical Biosciences and Engineering, 2019, 16(5): 4456-4476. doi: 10.3934/mbe.2019222

| [1] | J. Sivic and A. Zisserman, Video Google: A Text Retrieval Approach to Object Matching in Videos, Proc. Ninth International Conf. Computer Vision, 2003. |
| [2] | S. Lazebnik, C. Schmid and J. Ponce, Beyond bags of features: Spatial pyramid matching for recognizing natural scene categories, Computer vision and pattern recognition, New York, USA, 2006. |
| [3] | E. K. Wang, Y. P. Li, Y. M. Ye, et al., A Dynamic Trust Framework for Opportunistic Mobile Social Networks, IEEE T. Netw. Serv., 15 (2018), 319–329. |
| [4] | L. Gueguen, Classifying compound structures in satellite images: A compressed representation for fast queries, IEEE T. Geosci. Remote, 53 (2015), 1803–1818. |
| [5] | A. S. Razavian, H. Azizpour, J. Sullivan, et al., CNN features off-the-shelf: an astounding base-line for recognition,Proceedings of the IEEE conference on computer vision and pattern recogni-tion workshops. Columbus, OH, USA, (2014), 806–813. |
| [6] | M. Oquab, L. Bottou, I. Laptev, et al., Learning and transferring mid-level image representations using convolutional neural networks, Proceedings of the IEEE conference on computer vision and pattern recognition. Columbus, OH, USA, 2014. |
| [7] | R. Girshick, J. Donahue, T. Darrell, et al., Rich feature hierarchies for accurate object detection and semantic segmentation,Proceedings of the IEEE conference on computer vision and pattern recognition. Columbus, OH, USA, 2014. |
| [8] | J. Deng, W. Dong, R. Socher, et al., Imagenet: A large-scale hierarchical image database, IEEE Conference on Computer Vision and Pattern Recognition, 2009. |
| [9] | R. Salakhutdinov, J. B. Tenenbaum and A. Torralba, Learning with hierarchical-deep models, IEEE T. Pattern Anal., 35 (2013), 1958–1971. |
| [10] | F. Hu, G. S. Xia, J. Hu, et al., Transferring deep convolutional neural networks for the scene classification of high-resolution remote sensing imagery, Remote Sens-Basel, 7 (2015), 14680–14707. |
| [11] | W. Hu, Y. Huang, L. Wei, et al., Deep convolutional neural networks for hyperspectral image classification, J. Sensors, 15 (2015), 1–12. |
| [12] | F. Zhang, B. Du and L. Zhang, Saliency-guided unsupervised feature learning for scene classifi-cation, IEEE T. Geosci. Remote , 53 (2015), 2175–2184. |
| [13] | O. Firat, G. Can and F. T. Y. Vural, Representation learning for contextual object and region detection in remote sensing,Conference on Pattern Recognition (ICPR), 2014. |
| [14] | Y. Bengio, Deep Learning of Representations for Unsupervised and Transfer Learning, Interna-tional Conference on ICML Unsupervised and Transfer Learning, 2012. |
| [15] | G. Mesnil, Y. Dauphin, X. Glorot, et al., Unsupervised and Transfer Learning Challenge: a Deep Learning Approach, International Conference on ICML Unsupervised and Transfer Learning, 2012. |
| [16] | Y. LeCun, L. Bottou, G. B. Orr, et al., Efficient backprop.In Neural Networks: Tricks of the Trade, Springer, 1998. |
| [17] | A. M. Saxe, J. L. McClelland and S. Ganguli, Exact solutions to the nonlinear dynamics of learning in deep linear neural networks, arXiv preprint arXiv:1312.6120, 2013. |
| [18] | K.He, X.Zhang, S.Ren, et al., Delving deep into rectifiers: Surpassing human-level performance on imagenet classification, IEEE international conference on computer vision, 2015. |
| [19] | C. Szegedy, W. Liu, Y. Jia, et al., Going deeper with convolutions, IEEE Conference on Computer Vision and Pattern Recognition, 2015. |
| [20] | J. Wang, L. C. K. Hui, S. M. Yiu, et al., A survey on cyber attacks against nonlinear state estima-tion in power systems of ubiquitous cities, Pervasive Mob. Comput., 39 (2017), 10–17. |
| [21] | K. Chatfield, K. Simonyan, A. Vedaldi, et al., Return of the devil in the details: Delving deep into convolutional nets, arXiv preprint arXiv:1405.3531, 2014. |
| [22] | M. D. Zeiler and R. Fergus, Visualizing and understanding convolutional networks,European conference on computer vision, 2014. |
| [23] | S. Ioffe and C. Szegedy, Batch normalization: Accelerating deep network training by reducing internal covariate shift, International Conference on Machine Learning, 2015. |
| [24] | K. He, X. Zhang, S. Ren, et al., Deep residual learning for image recognition,IEEE conference on computer vision and pattern recognition, 2016. |
| [25] | Y. Yang and S. Newsam, Bag-of-visual-words and spatial extensions for land-use classification, Proceedings of the 18th SIGSPATIAL international conference on advances in geographic infor-mation systems. ACM, San Jose, CA, USA, (2010), 270–279. |
| [26] | G. S. Xia, W. Yang, J. Delon, et al., Structural high-resolution satellite image indexing, ISPRS TC VII Symposium-100 Years ISPRS, 2010. |
| [27] | O. Russakovsky, J. Deng, H. Su, et al., Imagenet large scale visual recognition challenge, Int. J. Comput. Vision, 115 (2017), 211–252. |
| [28] | C. M. Chen, B. Xiang, Y. Liu, et al., A Secure Authentication Protocol for Internet of Vehicles, IEEE Access, 7 (2019), 12047–12057. |
| [29] | K. H. Wang, C. M. Chen, W. C. Fang, et al., On the security of a new ultra-lightweight authenti-cation protocol in IoT environment for RFID tags, J. Supercomput., 74 (2018), 65–70. |
| [30] | E. Wang, Y. Li, Z. Nie, et al., Deep Fusion Feature Based Object Detection Method for High Resolution Optical Remote Sensing Images, Appl. Sci., 9 (2019), 1130–1148. |
| [31] | A. Karati, S. H. Islam and M. Karuppiah, Provably Secure and Lightweight Certificateless Sig-nature Scheme for IIoT Environments, IEEE T. Ind. Inform, 18 (2018), 1–8. |
| [32] | J. Guan and E. Wang, Repeated review based image captioning for image evidence review, Signal Process-Image, 63 (2018), 141–148. |




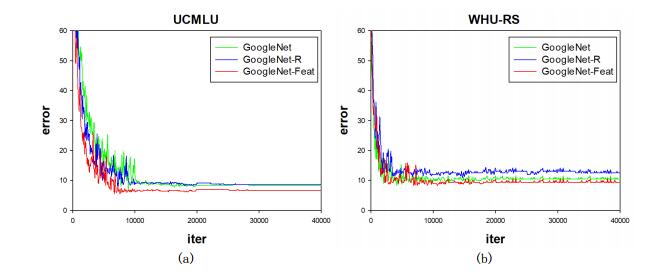
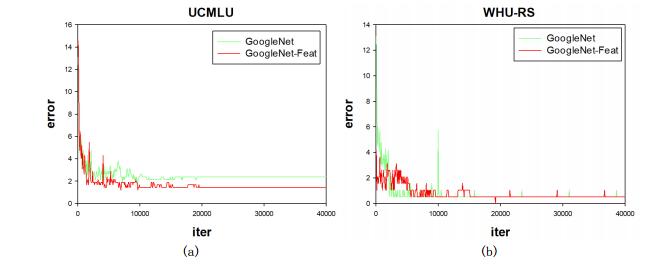
Figures(14) / Tables(7)

Eric Ke Wang, Fan Wang, Ruipei Sun, Xi Liu. A new privacy attack network for remote sensing images classification with small training samples[J]. Mathematical Biosciences and Engineering, 2019, 16(5): 4456-4476. doi: 10.3934/mbe.2019222







 DownLoad:
DownLoad: