Citation: Morteza Vaezi, G. Rezaei Behbehani, Nematollah Gheibi, Alireza Farasat. Thermodynamic, kinetic and docking studies of some unsaturated fatty acids-quercetin derivatives as inhibitors of mushroom tyrosinase[J]. AIMS Biophysics, 2020, 7(4): 393-410. doi: 10.3934/biophy.2020027

| [1] | Lind T, Siegbahn PEM, Crabtree RH (1999) A quantum chemical study of the mechanism of tyrosine. J Phys Chem B 103: 1193-1202. |
| [2] | Nokinsee D, Shank L, Lee VS, et al. (2015) Estimation of inhibitory effect against tyrosinase activity through homology modeling and molecular docking. Enzyme Res . |
| [3] | Ismaya WT, Rozeboom HJ, Weijn A, et al. (2011) Crystal structure of Agaricus bisporus mushroom tyrosinase: identity of the tetramer subunits and interaction with tropolone. Biochemistry 50: 5477-5486. |
| [4] | Matoba Y, Kihara S, Bando N, et al. (2018) Catalytic mechanism of the tyrosinase reaction toward the Tyr98 residue in the caddie protein. PLoS Biol 16: e3000077. |
| [5] | Murray AF (2016) Tyrosinase Inhibitors Identified from Phytochemicals and Their Mechanism of Control Berkeley. |
| [6] | Gou L, Lee J, Hao H, et al. (2017) The effect of oxaloacetic acid on tyrosinase activity and structure: Integration of inhibition kinetics with docking simulation. Int J Biol Macromol 101: 59-66. |
| [7] | Chang TS (2009) An updated review of tyrosinase inhibitors. Int J Mol Sci 10: 2440-2475. |
| [8] | Kim YJ, Uyama H (2005) Tyrosinase inhibitors from natural and synthetic sources: structure, inhibition mechanism and perspective for the future. Cell Mol Life Sci 62: 1707-1723. |
| [9] | Rho HS, Ahn SM, Lee BC, et al. (2010) Changes in flavonoid content and tyrosine inhibitory activity in kenaf leaf extract after far-infrared treatment. Bioorg Med Chem Lett 20: 7534-7536. |
| [10] | Da Hae G, Jo JM, Kim SY, et al. (2019) Tyrosinase inhibitors from natural source as skin-whitening agents and the application of edible insects: A mini review. Inter J Clin Nutr Diet 5. |
| [11] | Chang TS (2009) An updated review of tyrosinase inhibitors. J Molecul Sci 10: 2440-2475. |
| [12] | Glatz JFC, Börchers T, Spener F, et al. (1995) Fatty acids in cell signalling: modulation by lipid binding proteins. Prostag, Leukotr Ess 52: 121-127. |
| [13] | Mainini F, Contini A, Nava D, et al. (2013) Synthesis, molecular characterization and preliminary antioxidant activity evaluation of quercetin fatty esters. J Am Oil Chemists' Soc 90: 1751-1759. |
| [14] | Simopoulos AP (2002) Omega-3 fatty acids in inflammation and autoimmune diseases. J Am Coll Nutr 21: 495-505. |
| [15] | Johnson M, Bradford C (2014) Omega-3, omega-6 and omega-9 fatty acids: implications for cardiovascular and other diseases. J Glycomics Lipidomics 4: 2153-0637. |
| [16] | Ando H, Wen ZM, Kim HY, et al. (2006) Intracellular composition of fatty acid affects the processing and function of tyrosinase through the ubiquitin–proteasome pathway. Biochem J 394: 43-50. |
| [17] | Richards LB, Li M, van Esch BCAM, et al. (2016) The effects of short-chain fatty acids on the cardiovascular system. Pharma Nutrition 4: 68-111. |
| [18] | Khan F, Niaz K, Maqbool F, et al. (2016) Molecular targets underlying the anticancer effects of quercetin: an update. Nutrients 8: 529. |
| [19] | Warnakulasuriya SN, Rupasinghe HP (2014) Long chain fatty acid acylated derivatives of quercetin-3-O-glucoside as antioxidants to prevent lipid oxidation. Biomolecules 4: 980-993. |
| [20] | Jamali Z, Rezaei Behbehani G, Zare K, et al. (2019) Effect of chrysin omega-3 and 6 fatty acid esters on mushroom tyrosinase activity, stability, and structure. J Food Biochem 43: e12728. |
| [21] | Ashraf Z, Rafiq M, Seo SY, et al. (2015) Synthesis, kinetic mechanism and docking studies of vanillin derivatives as inhibitors of mushroom tyrosinase. Bioorgan Med Chem 23: 5870-5880. |
| [22] | Hassani S, Haghbeen K, Fazli M (2016) Non-specific binding sites help to explain mixed inhibition in mushroom tyrosinase activities. EurJ Med Chem 122: 138-148. |
| [23] | Li ZC, Chen LH, Yu XJ, et al. (2010) Inhibition kinetics of chlorobenzaldehyde thiosemicarbazones on mushroom tyrosinase. J Agr Food Chem 58: 12537-12540. |
| [24] | Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31: 455-461. |
| [25] | Matoba Y, Kihara S, Bando N, et al. (2018) Catalytic mechanism of the tyrosinase reaction toward the Tyr98 residue in the caddie protein. PLoS Biol 16: e3000077. |
| [26] | Studio A D (2006) 1.7 San Diego, CA, USA: Accelrys Software Inc.. |
| [27] | Mazhab-Jafari MT, Marshall CB, Smith MJ, et al. (2015) Oncogenic and RASopathy-associated K-RAS mutations relieve membrane-dependent occlusion of the effector-binding site. P Natl Acad Sci 112: 6625-6630. |
| [28] | Kusumaningrum S, Budianto E, Kosela S, et al. (2014) The molecular docking of 1, 4-naphthoquinone derivatives as inhibitors of Polo-like kinase 1 using Molegro Virtual Docker. J App Sci 4: 47-53. |
| [29] | Thomsen R, Christensen MH (2006) MolDock: a new technique for high-accuracy molecular docking. J Med Chem 49: 3315-3321. |
| [30] | Batra J Biophysical studies of protein folding and binding stability (2009) . |
| [31] | Gheibi N, Saboury AA, Haghbeen K, et al. (2009) Dual effects of aliphatic carboxylic acids on cresolase and catecholase reactions of mushroom tyrosinase. J Enzym Inhib Med Chem 24: 1076-1081. |
| [32] | Gheibi N, Zavareh SH, Behbahani GRR, et al. App Bioch Microbiol (2016) .52: 304-310. |
| [33] | Jamkhande PG, Ghante MH, Ajgunde BR (2017) Software based approaches for drug designing and development: a systematic review on commonly used software and its applications. Bulletin of Faculty of Pharmacy, Cairo University 55: 203-210. |
| [34] | Guo YJ, Pan ZZ, Chen CQ, et al. (2010) Inhibitory effects of fatty acids on the activity of mushroom tyrosinase. Appl Biochem Biotech 162: 1564-1573. |
| [35] | Lestari SR, Lukiati B, Arifah SN, et al. (2019) Medicinal uses of single garlic in hyperlipidemia by fatty acid synthase enzyme inhibitory: Molecular docking, IOP Conference Series: Earth and Environmental Science IOP Publishing, 012008. |
| [36] | Monserud JH, Schwartz DK (2012) Effects of molecular size and surface hydrophobicity on oligonucleotide interfacial dynamics. Biomacromolecules 13: 4002-4011. |
| [37] | Shalbafan M, Behbehani GR, Divsalar A (2018) The effect of methotrexate on the structural changes of human serum. J Ponte 74: 60-67. |
| [38] | Xue YL, Miyakawa T, Hayashi Y (2011) Isolation and tyrosinase inhibitory effects of polyphenols from the leaves of persimmon, Diospyros kaki. J Agr Food Chem 59: 6011-6017. |
| [39] | McDonnell JR, Reynolds RG, Fogel DB Docking conformationally flexible small molecules into a protein binding site through evolutionary programming (1995) . |
| [40] | Thomsen R, Christensen MH (2006) MolDock: A new technique for high-accuracy molecular docking. J Med Chem 49: 3315-3321. |
| [41] | Baek HS, Rho HS, Yoo JW, et al. (2008) The inhibitory effect of new hydroxamic acid derivatives on melanogenesis. B Korean Chem Soc 29: 43-46. |
| [42] | Kim YJ, Kang KS, Yokozawa T (2008) The anti-melanogenic effect of pycnogenol by its anti-oxidative actions. Food Chem Toxicol 46: 2466-2471. |
| [43] | Panich U, Pluemsamran T, Wattanarangsan J, et al. (2013) Protective effect of AVS073, a polyherbal formula, against UVA-induced melanogenesis through a redox mechanism involving glutathione-related antioxidant defense. BMC Complem Altern M 13: 1-10. |

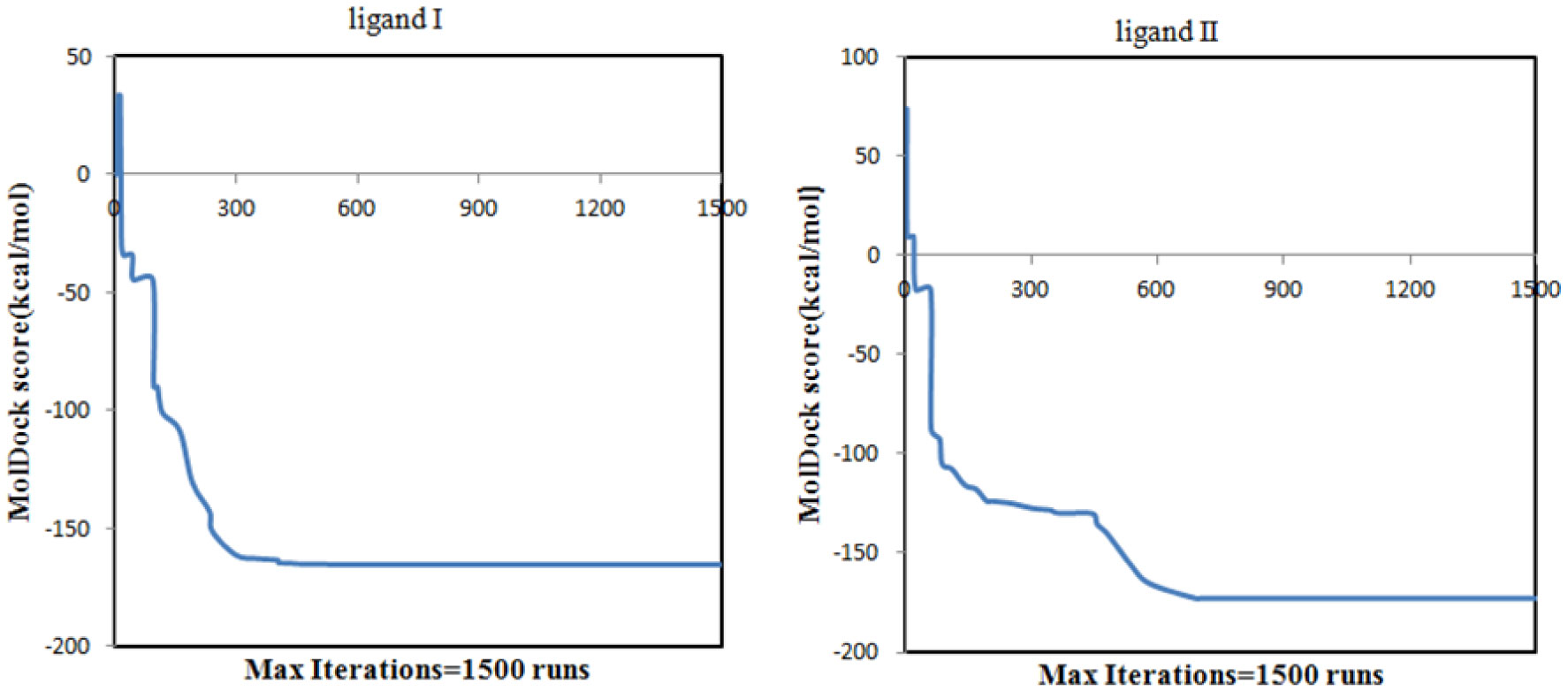
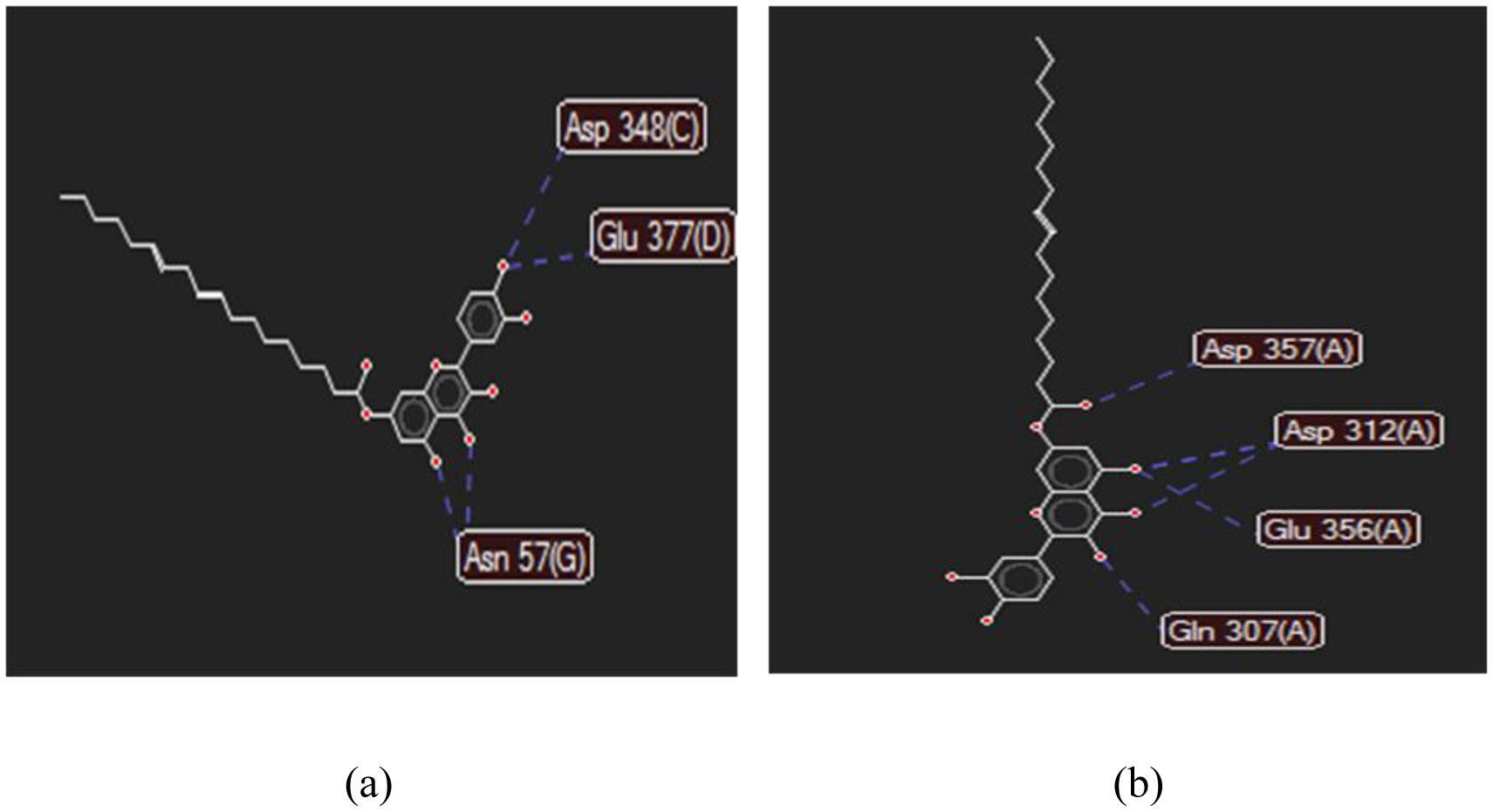
Figures(11) / Tables(4)

Morteza Vaezi, G. Rezaei Behbehani, Nematollah Gheibi, Alireza Farasat. Thermodynamic, kinetic and docking studies of some unsaturated fatty acids-quercetin derivatives as inhibitors of mushroom tyrosinase[J]. AIMS Biophysics, 2020, 7(4): 393-410. doi: 10.3934/biophy.2020027







 DownLoad:
DownLoad: