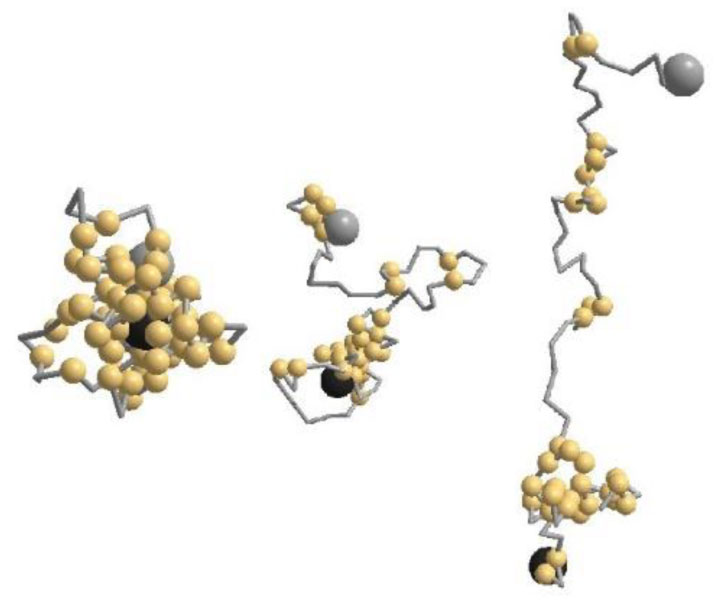
Thermal response of an envelope protein conformation from coronavirus-2 (CoVE) is studied by a coarse-grained Monte Carlo simulation. Three distinct segments, the N-terminal, Trans-membrane, and C-terminal are verified from its specific contact profile. The radius of gyration (Rg) reveals a non-monotonic sub-universal thermal response: Rg decays substantially on heating in native phase under low-temperature regime in contrast to a continuous increase on further raising the temperature prior to its saturation to a random-coil in denature phase. The globularity index which is a measure of effective dimension of the protein, decreases as the protein denatures from a globular to a random-coil conformation.
Citation: Panisak Boonamnaj, Pornthep Sompornpisut, Piyarat Nimmanpipug, R.B. Pandey. Thermal denaturation of a coronavirus envelope (CoVE) protein by a coarse-grained Monte Carlo simulation[J]. AIMS Biophysics, 2022, 9(4): 330-340. doi: 10.3934/biophy.2022027
Thermal response of an envelope protein conformation from coronavirus-2 (CoVE) is studied by a coarse-grained Monte Carlo simulation. Three distinct segments, the N-terminal, Trans-membrane, and C-terminal are verified from its specific contact profile. The radius of gyration (Rg) reveals a non-monotonic sub-universal thermal response: Rg decays substantially on heating in native phase under low-temperature regime in contrast to a continuous increase on further raising the temperature prior to its saturation to a random-coil in denature phase. The globularity index which is a measure of effective dimension of the protein, decreases as the protein denatures from a globular to a random-coil conformation.

| [1] |
Giri R, Bhardwaj T, Shegane M, et al. (2021) Understanding COVID‑19 via comparative analysis of dark proteomes of SARS‑CoV‑2, human SARS and bat SARS‑like coronaviruses. Cell Mol Life Sci 78: 1655-1688. https://doi.org/10.1007/s00018-020-03603-x 
|
| [2] |
Wan Y, Shang J, Graham R, et al. (2020) Receptor recognition by the novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J Virol 94: e00127-20. https://doi.org/10.1128/JVI.00127-20
|
| [3] |
Xu J, Zhao S, Teng T, et al. (2020) Systematic comparison of two animal-to-human transmitted human coronaviruses: SARS-CoV-2 and SARS-CoV. Viruses 12: 244. https://doi.org/10.3390/v12020244
|
| [4] |
Schoeman D, Fielding BC (2019) Coronavirus envelope protein: current knowledge. Virol J 16: 69. https://doi.org/10.1186/s12985-019-1182-0
|
| [5] |
Siu YL, Teoh KT, Lo J, et al. (2008) The M, E, and N structural proteins of the severe acute respiratory syndrome coronavirus are required for efficient assembly, trafficking, and release of virus-like particles. J Virol 82: 11318-11330. https://doi.org/10.1128/JVI.01052-08
|
| [6] |
Pawłowski PH (2021) Charged amino acids may promote coronavirus SARS-CoV-2 fusion with the host cell. AIMS Biophys 8: 111-120. https://doi.org/10.3934/biophy.2021008
|
| [7] | Pawłowski PH (2022) SARS-CoV-2 variant Omicron (B. 1.1. 529) is in a rising trend of mutations increasing the positive electric charge in crucial regions of the spike protein S. Acta Biochim Pol 69: 263-264. https://doi.org/10.18388/abp.2020_6072 |
| [8] | Wu C, Yin W, Jiang Y, et al. (2022) Structure genomics of SARS-CoV-2 and its Omicron variant: drug design templates for COVID-19. Acta Pharmacol Sin : 1-13. https://doi.org/10.1038/s41401-021-00851-w |
| [9] |
Naqvi AAT, Fatima K, Mohammad T, et al. (2020) Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: structural genomics approach. BBA-Mol Basis Dis 1866: 165878. https://doi.org/10.1016/j.bbadis.2020.165878
|
| [10] | Consortium UniProt (2021) UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res 49: D480-D489. https://doi.org/10.1093/nar/gkaa1100 |
| [11] |
Wilson L, Mckinlay C, Gage P, et al. (2004) SARS coronavirus E protein forms cation-selective ion channels. Virology 330: 322-331. https://doi.org/10.1016/j.virol.2004.09.033
|
| [12] |
Torres J, Wang J, Parthasarathy K, et al. (2005) The transmembrane oligomers of coronavirus protein E. Biophys J 88: 1283-1290. https://doi.org/10.1529/biophysj.104.051730
|
| [13] |
Liu J, Sun Y, Qi J, et al. (2010) The membrane protein of severe acute respiratory syndrome coronavirus acts as a dominant immunogen revealed by a clustering region of novel functionally and structurally defined cytotoxic T-lymphocyte epitopes. J Infect Dis 202: 1171-1180. https://doi.org/10.1086/656315
|
| [14] |
Venkatagopalan P, Daskalova SM, Lopez LA, et al. (2015) Coronavirus envelope(E) protein remains at the site of assembly. Virology 478: 75-85. https://doi.org/10.1016/j.virol.2015.02.005
|
| [15] |
Surya W, Li Y, Torres J (2018) Structural model of the SARS coronavirus E channel in LMPG micelles. BBA-Biomembranes 1860: 1309-1317. https://doi.org/10.1016/j.bbamem.2018.02.017
|
| [16] |
Gupta MK, Vemula S, Donde R, et al. (2021) In-silico approaches to detect inhibitors of the human severe acute respiratory syndrome coronavirus envelope protein ion channel. J Biomol Struct Dyn 39: 2617-2627. https://doi.org/10.1080/07391102.2020.1751300
|
| [17] |
Kuzmin A, Orekhov P, Astashkin R, et al. (2022) Structure and dynamics of the SARS-CoV-2 envelope protein monomer. Proteins 90: 1102-1114. https://doi.org/10.1002/prot.26317
|
| [18] | Binder K (1995) Monte Carlo and Molecular Dynamics Simulations in Polymer Science 1 Eds., New York: Oxford University Press. |
| [19] |
Fritsche M, Pandey RB, Farmer BL, et al. (2013) Variation in structure of a protein (H2AX) with knowledge-based interactions. PLoS One 8: e64507. https://doi.org/10.1371/journal.pone.0064507
|
| [20] |
Boonamnaj P, Paudel SS, Jetsadawisut W, et al. (2019) Thermal-response of a protein (hHv1) by a coarse-grained MC and all-atom MD computer simulations. Physica A 527: 121310. https://doi.org/10.1016/j.physa.2019.121310
|
| [21] |
Rangubpit W, Sompornpisut P, Pandey RB (2021) Thermal-induced unfolding-refolding of a nucleocapsid COVN protein. AIMS Biophys 8: 103-110. https://doi.org/10.3934/biophy.2021007
|
| [22] |
Betancourt MR, Thirumalai D (1999) Pair potentials for protein folding: choice of reference states and sensitivity of predicted native states to variations in the interaction schemes. Protein Sci 8: 361-369. https://doi.org/10.1110/ps.8.2.361
|
| [23] |
Miyazawa S, Jernigan RL (1985) Estimation of effective interresidue contact energies from protein crystal structures: quasi-chemical approximation. Macromolecules 18: 534-552. https://doi.org/10.1021/MA00145A039
|
| [24] |
Miyazawa S, Jernigan RL (1996) Residue-residue potentials with a favorable contact pair term and an unfavorable high packing density term, for simulation and treading. J Mol Biol 256: 623-644. https://doi.org/10.1006/JMBI.1996.0114
|
| [25] |
Tanaka S, Scheraga HA (1976) Medium- and long-range interaction parameters between amino acids for predicting three-dimensional structures of proteins. Macromolecules 9: 945-950. https://doi.org/10.1021/ma60054a013
|
| [26] | Godzik A (1996) >Knowledge-based potentials for protein folding: what can we learn from known protein structures? Structure 4: 363-366. https://doi.org/10.1016/s0969-2126(96)00041-x |
| [27] |
Huang SY, Zou X (2011) Statistical mechanics-based method to extract atomic distance-dependent potentials from protein structures. Proteins 79: 2648-2661. https://doi.org/10.1002/prot.23086
|
| [28] | Caetano DLZ, Metzler R, Cherstvy AG, et al. (2021) Adsorption of Lysozyme into a charged confining pore. https://doi.org/10.1101/2021.07.11.451934 |





Figures(6)

Panisak Boonamnaj, Pornthep Sompornpisut, Piyarat Nimmanpipug, R.B. Pandey. Thermal denaturation of a coronavirus envelope (CoVE) protein by a coarse-grained Monte Carlo simulation[J]. AIMS Biophysics, 2022, 9(4): 330-340. doi: 10.3934/biophy.2022027







 DownLoad:
DownLoad: