Citation: Warin Rangubpit, Sunan Kitjaruwankul, Panisak Boonamnaj, Pornthep Sompornpisut, R.B. Pandey. Globular bundles and entangled network of proteins (CorA) by a coarse-grained Monte Carlo simulation[J]. AIMS Biophysics, 2019, 6(2): 68-82. doi: 10.3934/biophy.2019.2.68

| [1] |
Furukawa Y, Nukina N (2013) Functional diversity of protein fibrillar aggregates from physiology to RNA granules to neurodegenerative diseases. Biochim Biophys Acta 1832: 1271–1278. doi: 10.1016/j.bbadis.2013.04.011

|
| [2] |
Bai Y, Luo Q, Liu J (2016) Protein self-assembly via supramolecular strategies. Chem Soc Rev 45: 2756–2767. doi: 10.1039/C6CS00004E
|
| [3] |
McManus JJ, Charbonneau P, Zaccarelli E, et al. (2016) The physics of protein self-assembly. Curr Opin Colloid Interface Sci 22: 73–79. doi: 10.1016/j.cocis.2016.02.011
|
| [4] |
Sgarbossa A (2012) Natural biomolecules protein aggregation: Emerging strategies against amyloidogenesis. Int J Mol Sci 13: 17121–17137. doi: 10.3390/ijms131217121
|
| [5] |
Sun H, Luo Q, Hou C, et al. (2017) Nanostructures based on protein self-assembly: From hierarchical construction to bioinspired materials. Nano Today 14: 16–41. doi: 10.1016/j.nantod.2017.04.006
|
| [6] |
Garcia-Seisdedos H, Empereur-Mot C, Elad N, et al. (2017) Proteins evolve on the edge of supramolecular self-assembly. Nature 548: 244–247. doi: 10.1038/nature23320
|
| [7] | Pandey RB, Farmer BL, Gerstman BS (2015) Self-assembly dynamics for the transition of a globular aggregate to a fibril network of lysozyme proteins via a coarse-grained Monte Carlo simulation. AIP Adv 5. |
| [8] |
Yang L, Liu A, Cao S, et al. (2016) Self-Assembly of proteins: Towards supramolecular materials. Chem Eur J 22: 15570–15582. doi: 10.1002/chem.201601943
|
| [9] |
Hmiel SP, Snavely MD, Florer JB, et al. (1989) Magnesium transport in Salmonella typhimurium: genetic characterization and cloning of three magnesium transport loci. J Bacteriol 171: 4742–4751. doi: 10.1128/jb.171.9.4742-4751.1989
|
| [10] | Maguire ME (1992) MgtA and MgtB: prokaryotic P-type ATPases that mediate Mg2+ influx. J Bioenerg Biomembr 24: 319–328. |
| [11] |
Kehres DG, Lawyer CH, Maguire ME (1998) The CorA magnesium transporter gene family. Microb Comp Genomics 3: 151–169. doi: 10.1089/omi.1.1998.3.151
|
| [12] | Eshaghi S, Niegowski D, Kohl A, et al. (2006) Crystal structure of a divalent metal ion transporter CorA at 2.9 angstrom resolution. Science 313: 354–357. |
| [13] |
Lunin VV, Dobrovetsky E, Khutoreskaya G, et al. (2006) Crystal structure of the CorA Mg2+ transporter. Nature 440: 833–837. doi: 10.1038/nature04642
|
| [14] |
Payandeh J, Li C, Ramjeesingh M, et al. (2008) Probing structure-function relationships and gating mechanisms in the CorA Mg2+ transport system. J Biol Chem 283: 11721–11733. doi: 10.1074/jbc.M707889200
|
| [15] |
Payandeh J, Pai EF (2006) A structural basis for Mg2+ homeostasis and the CorA translocation cycle. EMBO J 25: 3762–3773. doi: 10.1038/sj.emboj.7601269
|
| [16] |
Dalmas O, Cuello LG, Jogini V, et al. (2010) Structural Dynamics of the Magnesium-bound Conformation of CorA in a lipid bilayer. Structure 18: 868–878. doi: 10.1016/j.str.2010.04.009
|
| [17] |
Dalmas O, Sompornpisut P, Bezanilla F, et al. (2014) Molecular mechanism of Mg2+-dependent gating in CorA. Nat Commun 5: 3590. doi: 10.1038/ncomms4590
|
| [18] |
Neale C, Chakrabarti N, Pomorski P, et al. (2015) Hydrophobic gating of ion permeation in magnesium channel CorA. Plos Comput Biol 11: e1004303. doi: 10.1371/journal.pcbi.1004303
|
| [19] |
Kitjaruwankul S, Wapeesittipan P, Boonamnaj P, et al. (2016) Inner and outer coordination shells of Mg2+ in CorA selectivity filter from Molecular Dynamics simulations. J Phys Chem B 120: 406–417. doi: 10.1021/acs.jpcb.5b10925
|
| [20] |
Matthies D, Dalmas O, Borgnia MJ, et al. (2016) Cryo-EM structures of the magnesium channel CorA reveal symmetry break upon gating. Cell 164: 747–756. doi: 10.1016/j.cell.2015.12.055
|
| [21] |
Chakrabarti N, Neale C, Payandeh J, et al. (2010) An iris-like mechanism of pore dilation in the CorA magnesium transport system. Biophys J 98: 784–792. doi: 10.1016/j.bpj.2009.11.009
|
| [22] |
Nordin N, Guskov A, Phua T, et al. (2013) Exploring the structure and function of Thermotoga maritima CorA reveals the mechanism of gating and ion selectivity in Co2+/Mg2+ transport. Biochem J 451: 365–374. doi: 10.1042/BJ20121745
|
| [23] |
Kitjaruwankul S, Khrutto C, Sompornpisut P, et al. (2016) Asymmetry in structural response of inner and outer transmembrane segments of CorA protein by a coarse-grain model. J Chem Phys 145: 135101. doi: 10.1063/1.4963807
|
| [24] |
Kitjaruwankul S, Boonamnaj P, Paudel SS, et al. (2018) Thermal-induced folding and unfolding of a transmembrane protein (CorA). Physica A 506: 987–992. doi: 10.1016/j.physa.2018.05.014
|
| [25] |
Munishkina LA, Ahmad A, Fink AL, et al. (2008) Guiding protein aggregation with macromolecular crowding. Biochemistry 47: 8993–9006. doi: 10.1021/bi8008399
|
| [26] |
Minton AP (2001) The influence of macromolecular crowding and macromolecular confinement on biochemical reactions in physiological media. J Biol Chem 276: 10577–10580. doi: 10.1074/jbc.R100005200
|
| [27] |
Ellis RJ (2001) Macromolecular crowding: an important but neglected aspect of the intracellular environment. Curr Opin Struct Biol 11: 114–119. doi: 10.1016/S0959-440X(00)00172-X
|
| [28] |
Alas SJ, González-Pérez PP, Beltrán HI (2019) In silico minimalist approach to study 2D HP protein folding into an inhomogeneous space mimicking osmolyte effect: First trial in the search of foldameric backbones. BioSystems 181: 31–43. doi: 10.1016/j.biosystems.2019.04.005
|
| [29] |
González-Pérez PP, Orta DJ, Pena I, et al. (2017) A computational approach to studying protein folding problems considering the crucial role of the intracellular environment. J Comput Biol 24: 995–1013. doi: 10.1089/cmb.2016.0115
|
| [30] |
Tsao D, Dokholyan NV (2010) Macromolecular crowding induces polypeptide com paction and decreases folding cooperativity. Phys Chem Chem Phys 12: 3491–3500. doi: 10.1039/b924236h
|
| [31] |
Ping G, Yuan JM, Vallieres M, et al. (2003) Effects of confinement on protein folding and protein stability. J Chem Phys 118: 8042–8048. doi: 10.1063/1.1564053
|
| [32] |
Kuznetsova I, Zaslavsky B, Breydo L, et al. (2015) Beyond the excluded volume effects: mechanistic complexity of the crowded milieu. Molecules 20: 1377–1409. doi: 10.3390/molecules20011377
|
| [33] | Binder K (1995) Monte Carlo and Molecular Dynamics Simulations in Polymer Science. Oxford University Press. |
| [34] | Pandey RB, Farmer BL (2014) Aggregation and network formation in self-assembly of protein (H3.1) by a coarse-grained Monte Carlo simulation. J Chem Phys 141. |
| [35] | Betancourt MR, Thirumalai D. (1999) Pair potentials for protein folding: choice of reference states and sensitivity of predicted native states to variations in the interaction schemes. Protein Sci 2:361–369. |
| [36] |
Miyazawa S, Jernigan RL (1985) Estimation of effective inter residue contact energies from protein crystal structures: quasi-chemical approximation. Macromolecules 18:534–552. doi: 10.1021/ma00145a039
|
| [37] |
Miyazawa S, Jernigan RL (1996) Residue-residue potentials with a favorable contact pair term for simulation and treading. J Mol Biol 256: 623–644. doi: 10.1006/jmbi.1996.0114
|
| [38] |
Tanaka S, Scheraga HA. (1976) Medium and long range interaction parameters between amino acids for predicting three dimensional structures of proteins. Macromolecules 9: 945–950. doi: 10.1021/ma60054a013
|
| [39] |
Godzik A (1996) Knowledge-based potentials for protein folding: what can we learn from protein structures? Structure 4: 363–366. doi: 10.1016/S0969-2126(96)00041-X
|
| [40] |
Huang SY, Zou X. (2011) Statistical mechanics-based method to extract atomic distance-dependent potentials from protein structures. Proteins 79: 2648–2661. doi: 10.1002/prot.23086
|
| [41] |
Pandey RB, Kuang Z, Farmer BL, et al. (2012) Stability of peptide (P1, P2) binding to a graphene sheet via an all-atom to all-residue coarse-grained approach. Soft Matter 8: 9101–9109. doi: 10.1039/c2sm25870f
|
| [42] |
Feng J, Pandey RB, Berry RJ, et al. (2011) Adsorption mechanism of single amino acid and surfactant molecules to Au {111} surfaces in aqueous solution: design rules for metal binding molecules. Soft Matter 7: 2113–2120. doi: 10.1039/c0sm01118e
|
 biophy-06-02-068-s001.pdf biophy-06-02-068-s001.pdf |
 |
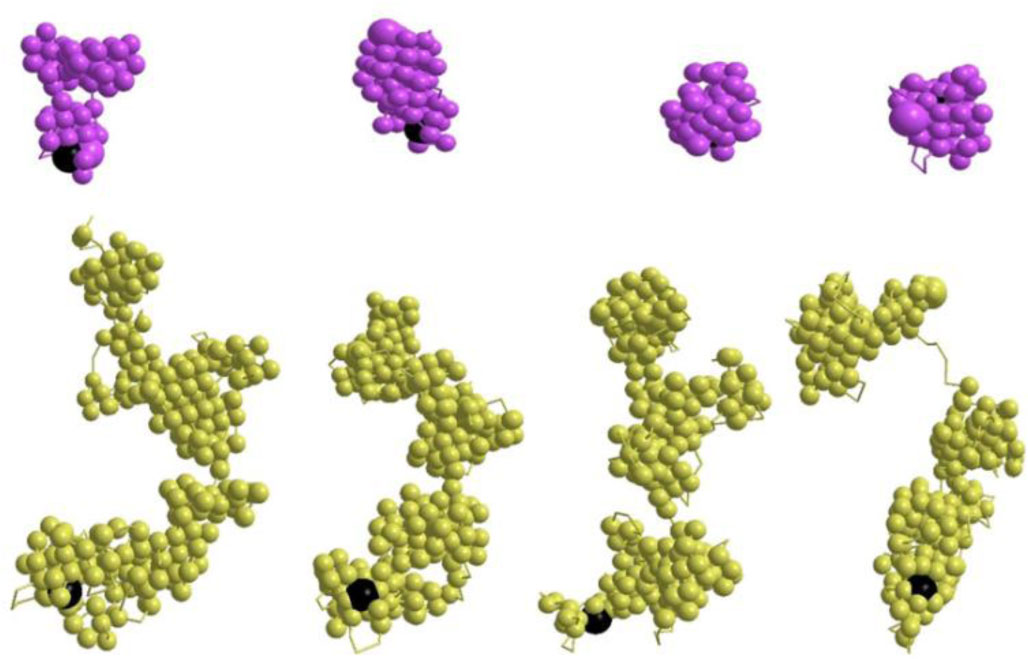
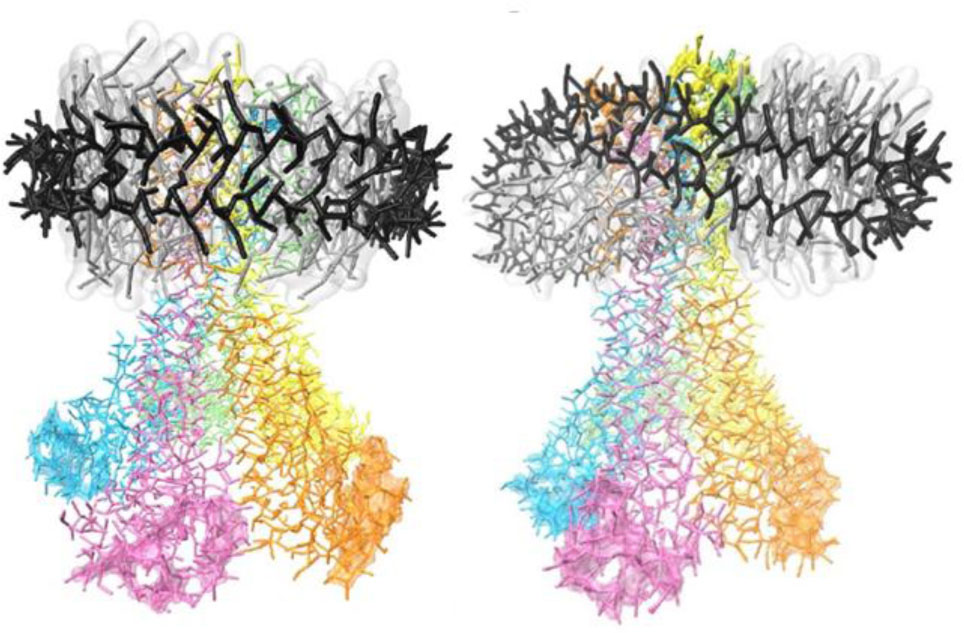
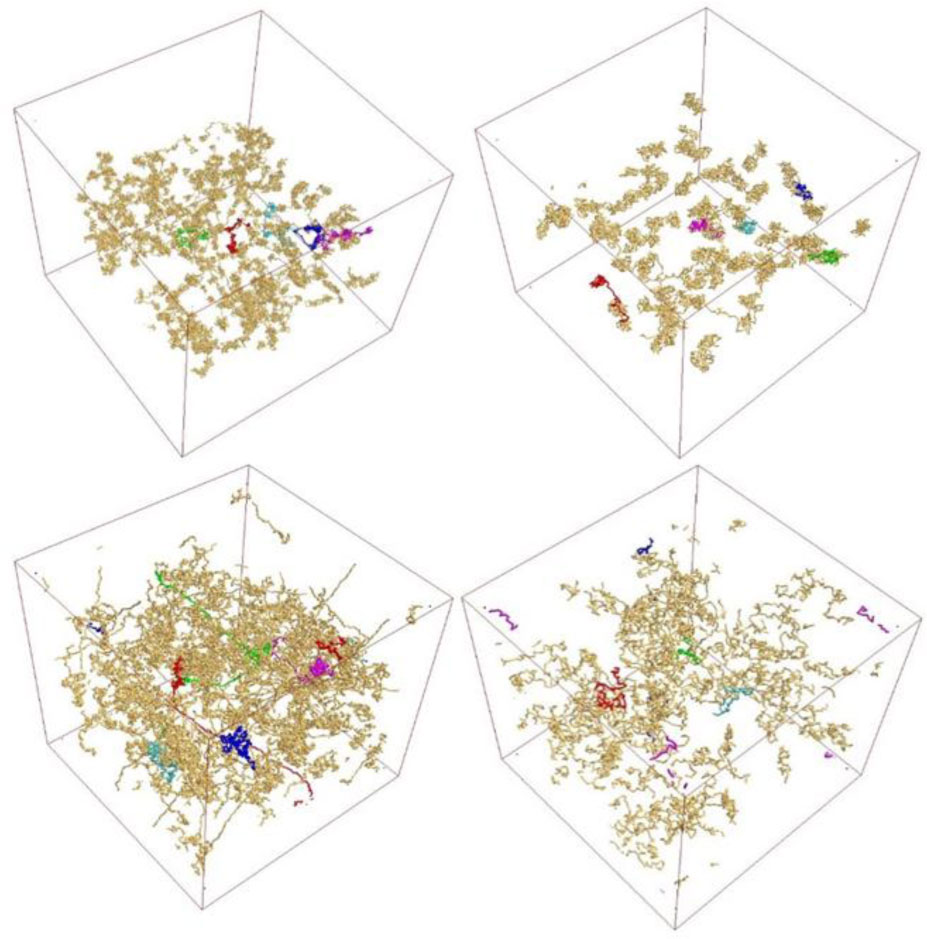
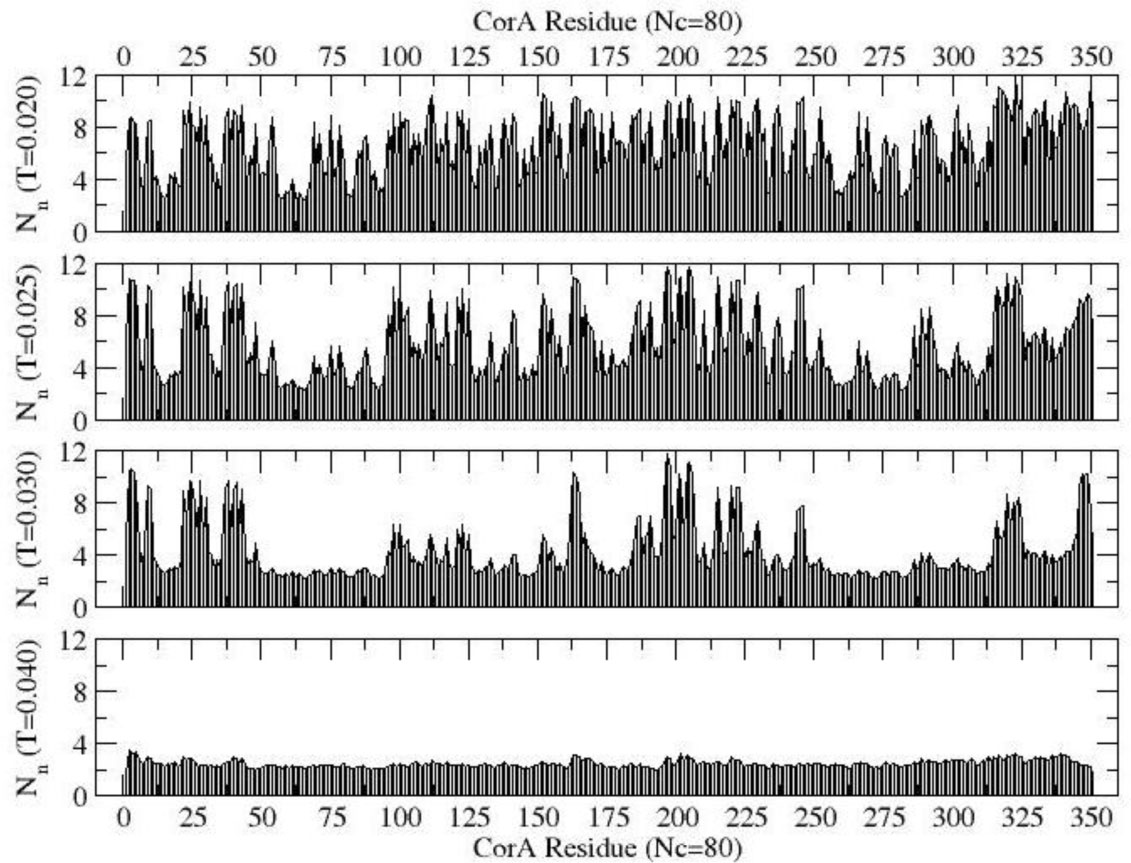
Figures(10)

Warin Rangubpit, Sunan Kitjaruwankul, Panisak Boonamnaj, Pornthep Sompornpisut, R.B. Pandey. Globular bundles and entangled network of proteins (CorA) by a coarse-grained Monte Carlo simulation[J]. AIMS Biophysics, 2019, 6(2): 68-82. doi: 10.3934/biophy.2019.2.68







 DownLoad:
DownLoad: