Citation: Nicole Lavender, David W. Hein, Guy Brock, La Creis R. Kidd. Evaluation of Oxidative Stress Response Related Genetic Variants, Pro-oxidants, Antioxidants and Prostate Cancer[J]. AIMS Medical Science, 2015, 2(4): 271-294. doi: 10.3934/medsci.2015.4.271

| [1] |
Sies H (1997) Oxidative stress: oxidants and antioxidants. Exp Physiol 82: 291-295. doi: 10.1113/expphysiol.1997.sp004024

|
| [2] |
Halliwell B (2007) Oxidative stress and cancer: have we moved forward? Biochem J 401: 1-11. doi: 10.1042/BJ20061131
|
| [3] |
Choi JY, Neuhouser ML, Barnett M, et al. (2007) Polymorphisms in oxidative stress-related genes are not associated with prostate cancer risk in heavy smokers. Cancer Epidemiol Biomarkers Prev 16: 1115-1120. doi: 10.1158/1055-9965.EPI-07-0040
|
| [4] |
Waris G, Ahsan H (2006) Reactive oxygen species: role in the development of cancer and various chronic conditions. J Carcinog 5: 14. doi: 10.1186/1477-3163-5-14
|
| [5] |
Miyake H, Hara I, Kamidono S, et al. (2004) Oxidative DNA damage in patients with prostate cancer and its response to treatment. J Urol 171: 1533-1536. doi: 10.1097/01.ju.0000116617.32728.ca
|
| [6] |
Khandrika L, Kumar B, Koul S, et al. (2009) Oxidative stress in prostate cancer. Cancer Lett 282: 125-136. doi: 10.1016/j.canlet.2008.12.011
|
| [7] |
Kumar B, Koul S, Khandrika L, et al. (2008) Oxidative stress is inherent in prostate cancer cells and is required for aggressive phenotype. Cancer Res 68: 1777-1785. doi: 10.1158/0008-5472.CAN-07-5259
|
| [8] |
Pathak SK, Sharma RA, Steward WP, et al. (2005) Oxidative stress and cyclooxygenase activity in prostate carcinogenesis: targets for chemopreventive strategies. Eur J Cancer 41: 61-70. doi: 10.1016/j.ejca.2004.09.028
|
| [9] |
Caceres DD, Iturrieta J, Acevedo C, et al. (2005) Relationship among metabolizing genes, smoking and alcohol used as modifier factors on prostate cancer risk: exploring some gene-gene and gene-environment interactions. Eur J Epidemiol 20: 79-88. doi: 10.1007/s10654-004-1632-9
|
| [10] |
Cross AJ, Peters U, Kirsh VA, et al. (2005) A prospective study of meat and meat mutagens and prostate cancer risk. Cancer Res 65: 11779-11784. doi: 10.1158/0008-5472.CAN-05-2191
|
| [11] |
Fleshner NE, Klotz LH (1998) Diet, androgens, oxidative stress and prostate cancer susceptibility. Cancer Metastasis Rev 17: 325-330. doi: 10.1023/A:1006118628183
|
| [12] |
Gong Z, Agalliu I, Lin DW, et al. (2008) Cigarette smoking and prostate cancer-specific mortality following diagnosis in middle-aged men. Cancer Causes Control 19: 25-31. doi: 10.1007/s10552-007-9066-9
|
| [13] |
Huncharek M, Haddock KS, Reid R, et al. (2010) Smoking as a risk factor for prostate cancer: a meta-analysis of 24 prospective cohort studies. Am J Public Health 100: 693-701. doi: 10.2105/AJPH.2008.150508
|
| [14] |
Kolonel LN (2001) Fat, meat, and prostate cancer. Epidemiol Rev 23: 72-81. doi: 10.1093/oxfordjournals.epirev.a000798
|
| [15] |
Koutros S, Berndt SI, Sinha R, et al. (2009) Xenobiotic metabolizing gene variants, dietary heterocyclic amine intake, and risk of prostate cancer. Cancer Res 69: 1877-1884. doi: 10.1158/0008-5472.CAN-08-2447
|
| [16] |
Rohrmann S, Genkinger JM, Burke A, et al. (2007) Smoking and risk of fatal prostate cancer in a prospective U.S. study. Urology 69: 721-725. doi: 10.1016/j.urology.2006.12.020
|
| [17] |
Sinha R, Park Y, Graubard BI, et al. (2009) Meat and meat-related compounds and risk of prostate cancer in a large prospective cohort study in the United States. Am J Epidemiol 170: 1165-1177. doi: 10.1093/aje/kwp280
|
| [18] |
Lotufo PA, Lee IM, Ajani UA, et al. (2000) Cigarette smoking and risk of prostate cancer in the physicians' health study (United States). Int J Cancer 87: 141-144. doi: 10.1002/1097-0215(20000701)87:1<141::AID-IJC21>3.0.CO;2-A
|
| [19] |
Watters JL, Park Y, Hollenbeck A, et al. (2009) Cigarette smoking and prostate cancer in a prospective US cohort study. Cancer Epidemiol Biomarkers Prev 18: 2427-2435. doi: 10.1158/1055-9965.EPI-09-0252
|
| [20] | Boelsterli UA (2007) Mechanistic Toxicology: the molecular basis of how chemicals disrupt biological targets. Boca Raton, FL: CRC Press. |
| [21] |
Cross AJ, Sinha R (2004) Meat-related mutagens/carcinogens in the etiology of colorectal cancer. Environ Mol Mutagen 44: 44-55. doi: 10.1002/em.20030
|
| [22] |
Kushi LH, Byers T, Doyle C, et al. (2006) American Cancer Society Guidelines on Nutrition and Physical Activity for cancer prevention: reducing the risk of cancer with healthy food choices and physical activity. CA Cancer J Clin 56: 254-281. doi: 10.3322/canjclin.56.5.254
|
| [23] |
Chan R, Lok K, Woo J (2009) Prostate cancer and vegetable consumption. Mol Nutr Food Res 53: 201-216. doi: 10.1002/mnfr.200800113
|
| [24] |
Ma RW, Chapman K (2009) A systematic review of the effect of diet in prostate cancer prevention and treatment. J Hum Nutr Diet 22: 187-199. doi: 10.1111/j.1365-277X.2009.00946.x
|
| [25] |
Kovacic P, Jacintho JD (2001) Mechanisms of carcinogenesis: focus on oxidative stress and electron transfer. Curr Med Chem 8: 773-796. doi: 10.2174/0929867013373084
|
| [26] |
Mates JM, Perez-Gomez C, Nunez dCI (1999) Antioxidant enzymes and human diseases. Clin Biochem 32: 595-603. doi: 10.1016/S0009-9120(99)00075-2
|
| [27] |
Aydin A, rsova-Sarafinovska Z, Sayal A, et al. (2006) Oxidative stress and antioxidant status in non-metastatic prostate cancer and benign prostatic hyperplasia. Clin Biochem 39: 176-179. doi: 10.1016/j.clinbiochem.2005.11.018
|
| [28] | National Center for Biotechnology Information (NCBI) website (2011). |
| [29] | Gamage N, Barnett A, Hempel N, et al. (2006) Human Sulfotransferases and Their Role in Chemical Metabolism. Toxicolog Sci 90: 5-22. |
| [30] |
Hein DW (2002) Molecular genetics and function of NAT1 and NAT2: role in aromatic amine metabolism and carcinogenesis. Mutat Res 506-507: 65-77. doi: 10.1016/S0027-5107(02)00153-7
|
| [31] |
Gross GA, Turesky RJ, Fay LB, et al. (1993) Heterocyclic aromatic amine formation in grilled bacon, beef and fish and in grill scrapings. Carcino Genesis 14: 2313-2318. doi: 10.1093/carcin/14.11.2313
|
| [32] | Badawi AF, Hirvonen A, Bell DA, et al. (1995) Role of aromatic amine acetyltransferases, NAT1 and NAT2, in carcinogen-DNA adduct formation in the human urinary bladder. Cancer Res 55: 5230-5237. |
| [33] |
Sikka SC (2003) Role of oxidative stress response elements and antioxidants in prostate cancer pathobiology and chemoprevention--a mechanistic approach. Curr Med Chem 10: 2679-2692. doi: 10.2174/0929867033456341
|
| [34] |
Zhou SF, Wang B, Yang LP, et al. (2010) Structure, function, regulation and polymorphism and the clinical significance of human cytochrome P450 1A2. Drug Metab Rev 42: 268-354. doi: 10.3109/03602530903286476
|
| [35] |
Metry KJ, Neale JR, Doll MA, et al. (2010) Effect of rapid human N-acetyltransferase 2 haplotype on DNA damage and mutagenesis induced by 2-amino-3-methylimidazo-[4,5-f]quinoline (IQ) and 2-amino-3,8-dimethylimidazo-[4,5-f]quinoxaline (MeIQx). Mutat Res 684: 66-73. doi: 10.1016/j.mrfmmm.2009.12.001
|
| [36] |
Autrup JL, Thomassen LH, Olsen JH, et al. (1999) Glutathione S-transferases as risk factors in prostate cancer. Eur J Cancer Prev 8: 525-532. doi: 10.1097/00008469-199912000-00008
|
| [37] |
Beer TM, Evans AJ, Hough KM, et al. (2002) Polymorphisms of GSTP1 and related genes and prostate cancer risk. Prostate Cancer Prostatic Dis 5: 22-27. doi: 10.1038/sj.pcan.4500549
|
| [38] |
Gsur A, Haidinger G, Hinteregger S, et al. (2001) Polymorphisms of glutathione-S-transferase genes (GSTP1, GSTM1 and GSTT1) and prostate-cancer risk. Int J Cancer 95: 152-155. doi: 10.1002/1097-0215(20010520)95:3<152::AID-IJC1026>3.0.CO;2-S
|
| [39] | Wadelius M, Autrup JL, Stubbins MJ, et al. (1999) Polymorphisms in NAT2, CYP2D6, CYP2C19 and GSTP1 and their association with prostate cancer. Pharmaco genetics 9: 333-340. |
| [40] |
Forsberg L, de FU, Morgenstern R (2001) Oxidative stress, human genetic variation, and disease. Arch Biochem Biophys 389: 84-93. doi: 10.1006/abbi.2001.2295
|
| [41] |
Kang D, Lee KM, Park SK, et al. (2007) Functional variant of manganese superoxide dismutase (SOD2 V16A) polymorphism is associated with prostate cancer risk in the prostate, lung, colorectal, and ovarian cancer study. Cancer Epidemiol Biomarkers Prev 16: 1581-1586. doi: 10.1158/1055-9965.EPI-07-0160
|
| [42] | U.S. Human and Health Services (2005) 11th Report on Carcinogens. |
| [43] |
Zheng W, Lee SA (2009) Well-done meat intake, heterocyclic amine exposure, and cancer risk. Nutr Cancer 61: 437-446. doi: 10.1080/01635580802710741
|
| [44] | Cancer Genetic Markers of Susceptibility (CGEMS) (2008). |
| [45] |
Gohagan JK, Prorok PC, Hayes RB, et al. (2000) The Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Trial of the National Cancer Institute: history, organization, and status. Control Clin Trials 21: 251S-272S. doi: 10.1016/S0197-2456(00)00097-0
|
| [46] |
Hayes RB, Sigurdson A, Moore L, et al. (2005) Methods for etiologic and early marker investigations in the PLCO trial. Mutat Res 592: 147-154. doi: 10.1016/j.mrfmmm.2005.06.013
|
| [47] |
Hasson MA, Fagerstrom RM, Kahane DC, et al. (2000) Design and evolution of the data management systems in the Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Trial. Control Clin Trials 21: 329S-348S. doi: 10.1016/S0197-2456(00)00100-8
|
| [48] | US Department of Health and Human Services and US Department of Agriculture (2005) Dietary Guidelines for Americans. 6th ed. Washington, DC: US Government Printing Office. |
| [49] | National Institutes of Health Office of Dietary Supplements. |
| [50] | Kanehisa M, Araki M, Goto S, et al. (2008) KEGG for linking genomes to life and the environment. Nucleic Acids Res 36: D480-D484. |
| [51] |
Kanehisa M, Goto S, Hattori M, et al. (2006) From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res 34: D354-D357. doi: 10.1093/nar/gkj102
|
| [52] |
Kanehisa M, Goto S (2000) KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28: 27-30. doi: 10.1093/nar/28.1.27
|
| [53] | Ingenuity Systems (2010) Ingenuity Pathways Analysis. |
| [54] | BioCarta LLC (2009) BioCarta.com |
| [55] |
Yue P, Melamud E, Moult J (2006) SNPs3D: candidate gene and SNP selection for association studies. BMC Bioinform 7: 166. doi: 10.1186/1471-2105-7-166
|
| [56] |
Xu Z, Taylor JA (2009) SNPinfo: integrating GWAS and candidate gene information into functional SNP selection for genetic association studies. Nucleic Acids Res 37: W600-W605. doi: 10.1093/nar/gkp290
|
| [57] |
Ramensky V, Bork P, Sunyaev S (2002) Human non-synonymous SNPs: server and survey. Nucleic Acids Res 30: 3894-3900. doi: 10.1093/nar/gkf493
|
| [58] | Menashe I, Rosenberg PS, Chen BE (2008) PGA: power calculator for case-control genetic association analyses. BMC Genet 9: 36. |
| [59] |
Moore JH, Gilbert JC, Tsai CT, et al. (2006) A flexible computational framework for detecting, characterizing, and interpreting statistical patterns of epistasis in genetic studies of human disease susceptibility. J Theor Biol 241: 252-261. doi: 10.1016/j.jtbi.2005.11.036
|
| [60] | Andrew AS, Nelson HH, Kelsey KT, et al. (2005) Concordance of multiple analytical approaches demonstrates a complex relationship between DNA repair gene SNPs, smoking, and bladder cancer susceptibility. Carcino Genesis: 1030-1037. |
| [61] |
Hahn LW, Ritchie MD, Moore JH (2003) Multifactor dimensionality reduction software for detecting gene-gene and gene-environment interactions. Bioinformatics 19: 376-382. doi: 10.1093/bioinformatics/btf869
|
| [62] |
Moore JH (2004) Computational analysis of gene-gene interactions using multifactor dimensionality reduction. Expert Rev Mol Diagn 4: 795-803. doi: 10.1586/14737159.4.6.795
|
| [63] | Gui J, Andrew AS, Andrews P, et al. (2010) A Robust Multifactor Dimensionality Reduction Method for Detecting Gene-Gene Interactions with Application to the Genetic Analysis of Bladder Cancer Susceptibility. Ann Hum Genet. |
| [64] |
Ritchie MD, Hahn LW, Moore JH (2003) Power of multifactor dimensionality reduction for detecting gene-gene interactions in the presence of genotyping error, missing data, phenocopy, and genetic heterogeneity. Genet Epidemiol 24: 150-157. doi: 10.1002/gepi.10218
|
| [65] | Jakulin A, Bratko I (2003) Analyzing attribute depedencies.In Lavrac N , Gamberger D, Blockeel H and Todorovski L (eds.)PKDD 2003. Cavtat, Croatia.: Springer-Verlag. pp. 229-240-240. |
| [66] | Jakulin A, Bratko I, Smrike D, et al. (2003) Attribute interactions in medical data analysis. Protarus: Cyprus. pp. 229-238-238. |
| [67] | Demosar J, Zupan B (2004) Orange: From Experimental Machine Learning to Interactive Data Mining, White Paper. |
| [68] |
Coates PJ, Lorimore SA, Wright EG (2005) Cell and tissue responses to genotoxic stress. J Pathol 205: 221-235. doi: 10.1002/path.1701
|
| [69] |
Hein DW, Fretland AJ, Doll MA (2006) Effects of single nucleotide polymorphisms in human N-acetyltransferase 2 on metabolic activation (O-acetylation) of heterocyclic amine carcinogens. Int J Cancer 119: 1208-1211. doi: 10.1002/ijc.21957
|
| [70] |
Zang Y, Doll MA, Zhao S, et al. (2007) Functional characterization of single-nucleotide polymorphisms and haplotypes of human N-acetyltransferase 2. Carcino Genesis 28: 1665-1671. doi: 10.1093/carcin/bgm085
|
| [71] |
Agundez JA, Olivera M, Ladero JM, et al. (1996) Increased risk for hepatocellular carcinoma in NAT2-slow acetylators and CYP2D6-rapid metabolizers. Pharmacogenetics 6: 501-512. doi: 10.1097/00008571-199612000-00003
|
| [72] |
Agundez JA, Olivera M, Martinez C, et al. (1996) Identification and prevalence study of 17 allelic variants of the human NAT2 gene in a white population. Pharmacogenetics 6: 423-428. doi: 10.1097/00008571-199610000-00006
|
| [73] | Anitha A, Banerjee M (2003) Arylamine N-acetyltransferase 2 polymorphism in the ethnic populations of South India. Int J Mol Med 11: 125-131. |
| [74] |
Patin E, Barreiro LB, Sabeti PC, et al. (2006) Deciphering the ancient and complex evolutionary history of human arylamine N-acetyltransferase genes. Am J Hum Genet 78: 423-436. doi: 10.1086/500614
|
| [75] |
Woolhouse NM, Qureshi MM, Bayoumi RA (1997) A new mutation C759T in the polymorphic N-acetyltransferase (NAT2) gene. Pharmacogenetics 7: 83-84. doi: 10.1097/00008571-199702000-00011
|
| [76] |
Sharma S, Cao X, Wilkens LR, et al. (2010) Well-done meat consumption, NAT1 and NAT2 acetylator genotypes and prostate cancer risk: the multiethnic cohort study. Cancer Epidemiol Biomarkers Prev 19: 1866-1870. doi: 10.1158/1055-9965.EPI-10-0231
|
| [77] |
Nock NL, Tang D, Rundle A, et al. (2007) Associations between smoking, polymorphisms in polycyclic aromatic hydrocarbon (PAH) metabolism and conjugation genes and PAH-DNA adducts in prostate tumors differ by race. Cancer Epidemiol Biomarkers Prev 16: 1236-1245. doi: 10.1158/1055-9965.EPI-06-0736
|
| [78] |
Koutros S, Andreotti G, Berndt SI, et al. (2011) Xenobiotic-metabolizing gene variants, pesticide use, and the risk of prostate cancer. Pharmacogenet Genomics 21: 615-623. doi: 10.1097/FPC.0b013e3283493a57
|
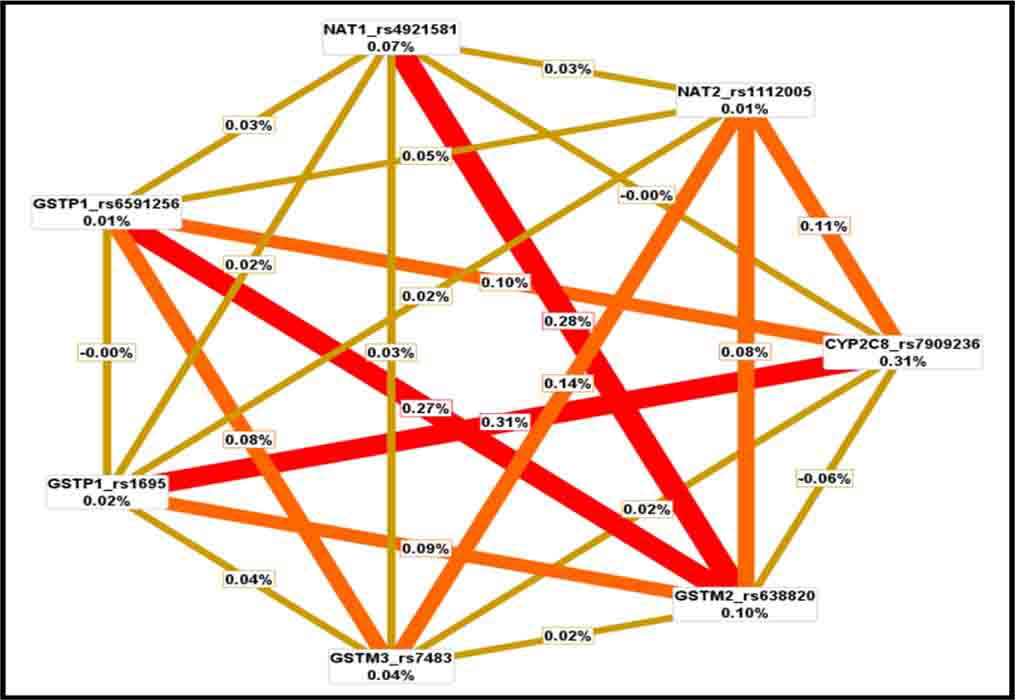
Figures(1) / Tables(10)

Nicole Lavender, David W. Hein, Guy Brock, La Creis R. Kidd. Evaluation of Oxidative Stress Response Related Genetic Variants, Pro-oxidants, Antioxidants and Prostate Cancer[J]. AIMS Medical Science, 2015, 2(4): 271-294. doi: 10.3934/medsci.2015.4.271







 DownLoad:
DownLoad: