Genito-pelvic pain penetration disorders involve a variety of sexual disorders associated with persistent pelvic pain, among which vulvodynia/vestibulodynia, dyspareunia, and vaginismus are usually found. The purpose of the current systematic review is to examine the efficacy of Transcutaneous Electrical Nerve Stimulation in women with genito-pelvic pain penetration disorders.
A wide search of the literature was performed for articles indexed on PubMed, Scopus, Web of Science, and Science Direct. This systematic review was registered on the International Prospective Register of Systematic Reviews (PROSPERO) database (RD42023443931). It was reported according to the Preferred Reporting Items for Systematic Reviews and Meta-Analysis standards.
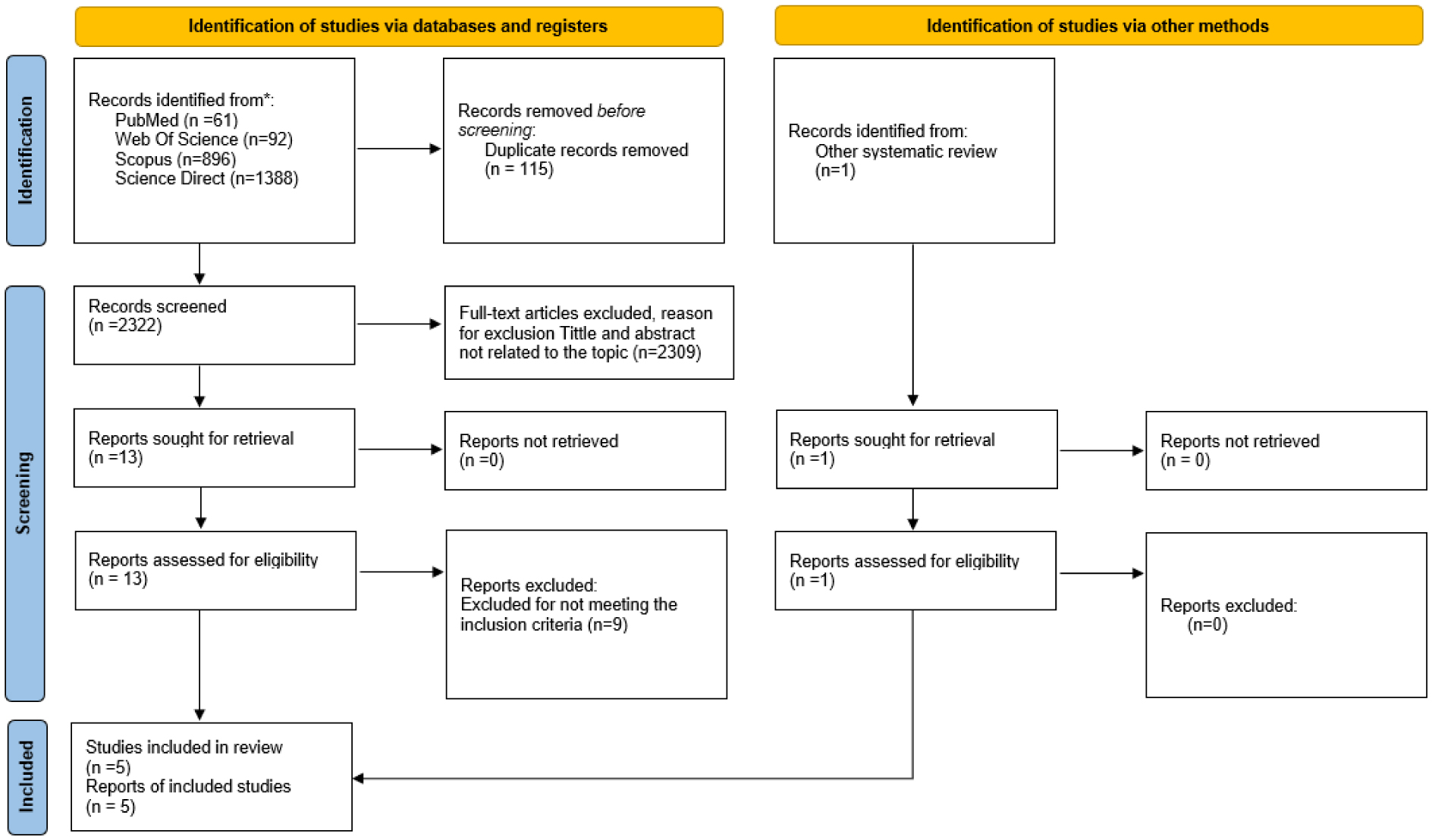
A total of five studies with 208 women with genito-pelvic pain penetration disorders were included. Transcutaneous Electrical Nerve Stimulation was applied either isolated or combined with other treatments, such as manual intravaginal techniques, pelvic floor muscle exercises, or pharmacological treatments. The number of sessions of the treatment ranged from 8 to 24 sessions, and the duration of the applied stimulus varied from 20 min to 30 min. After the intervention, pain, dyspareunia severity, the strength and endurance of pelvic floor muscles, and sexual function significantly improved in the experimental group, and at the 3 months follow-up.
Transcutaneous Electrical Nerve Stimulation improved the pain, dyspareunia severity, strength and endurance of pelvic floor muscles, and sexual function at the end of the intervention and at the 3 months follow up in patients with genito-pelvic pain penetration disorders. The use of additional treatments or techniques could also be beneficial in the treatment of these women due to the multifactorial origin of the disorder.
Citation: López-López Laura, Villalobos-Santos Lourdes, Del Castillo-Matías Rocío, Torres-Sánchez Irene, Díaz-Mohedo Esther. Transcutaneous electrical nerve stimulation in the treatment of women with genito-pelvic pain penetration disorders: a systematic review[J]. AIMS Medical Science, 2024, 11(3): 348-360. doi: 10.3934/medsci.2024024
Genito-pelvic pain penetration disorders involve a variety of sexual disorders associated with persistent pelvic pain, among which vulvodynia/vestibulodynia, dyspareunia, and vaginismus are usually found. The purpose of the current systematic review is to examine the efficacy of Transcutaneous Electrical Nerve Stimulation in women with genito-pelvic pain penetration disorders.
A wide search of the literature was performed for articles indexed on PubMed, Scopus, Web of Science, and Science Direct. This systematic review was registered on the International Prospective Register of Systematic Reviews (PROSPERO) database (RD42023443931). It was reported according to the Preferred Reporting Items for Systematic Reviews and Meta-Analysis standards.
A total of five studies with 208 women with genito-pelvic pain penetration disorders were included. Transcutaneous Electrical Nerve Stimulation was applied either isolated or combined with other treatments, such as manual intravaginal techniques, pelvic floor muscle exercises, or pharmacological treatments. The number of sessions of the treatment ranged from 8 to 24 sessions, and the duration of the applied stimulus varied from 20 min to 30 min. After the intervention, pain, dyspareunia severity, the strength and endurance of pelvic floor muscles, and sexual function significantly improved in the experimental group, and at the 3 months follow-up.
Transcutaneous Electrical Nerve Stimulation improved the pain, dyspareunia severity, strength and endurance of pelvic floor muscles, and sexual function at the end of the intervention and at the 3 months follow up in patients with genito-pelvic pain penetration disorders. The use of additional treatments or techniques could also be beneficial in the treatment of these women due to the multifactorial origin of the disorder.

| [1] | Vicente-Neira A, Prieto-Gómez V, Navarro-Brazález B, et al. (2022) Online information on painful sexual dysfunction in women: quality analysis of websites in SPANISH about dyspareunia, vaginismus and vulvodynia. Int J Environ Res Public Health 19: 1506. https://doi.org/10.3390/ijerph19031506 |
| [2] | (2022) American Psychiatric AssociationDiagnostic and statistical manual of mental disorders (5th ed., text rev.). American Psychiatric Publishing. |
| [3] | da Silva Nunes AC, Lemos CIL, dos Santos Araújo N, et al. (2019) Physiotherapeutic approach in genito-pelvic pain/penetration disorder. Man Ther Posturology Rehabil J 17: 707. https://doi.org/10.17784/mtprehabjournal.2019.17.707 |
| [4] | Stockdale CK, Lawson HW (2014) 2013 Vulvodynia guideline update. J Low Genit Tract Dis 18: 93-100. https://doi.org/10.1097/LGT.0000000000000021 |
| [5] | Reed BD, Harlow SD, Sen A, et al. (2012) Prevalence and demographic characteristics of vulvodynia in a population-based sample. Am J Obstet Gynecol 206: 170.e1-170.e9. https://doi.org/10.1016/j.ajog.2011.08.012 |
| [6] | Van Lankveld JJDM, Granot M, Weijmar Schultz WCM, et al. (2010) Women's sexual pain disorders. J Sex Med 7: 615-631. https://doi.org/10.1111/j.1743-6109.2009.01631.x |
| [7] | Berghmans B (2018) Physiotherapy for pelvic pain and female sexual dysfunction: an untapped resource. Int Urogynecol J 29: 631-638. https://doi.org/10.1007/s00192-017-3536-8 |
| [8] | Al-Abbadey M, Liossi C, Curran N, et al. (2015) Treatment of female sexual pain disorders: a systematic review. J Sex Marital Ther 42: 99-142. https://doi.org/10.1080/0092623X.2015.1053023 |
| [9] | Rosenbaum TY (2007) Physical therapy management and treatment of sexual pain disorders. Principles and Practice of Sex Therapy . New York: Guildford Press 157-177. |
| [10] | Gbiri CAO, Akumabor JC (2023) Effectiveness of physiotherapy interventions in the management male sexual dysfunction: a systematic review. Int J Sex Health 35: 52-66. https://doi.org/10.1080/19317611.2022.2155288 |
| [11] | Fernández-Pérez P, Leirós-Rodríguez R, Marqués-Sánchez MP, et al. (2023) Effectiveness of physical therapy interventions in women with dyspareunia: a systematic review and meta-analysis. BMC Womens Health 23: 387. https://doi.org/10.1186/s12905-023-02532-8 |
| [12] | Johnson MI, Paley CA, Jones G, et al. (2022) Efficacy and safety of transcutaneous electrical nerve stimulation (TENS) for acute and chronic pain in adults: a systematic review and meta-analysis of 381 studies (the meta-TENS study). BMJ Open 12: e051073. https://doi.org/10.1136/bmjopen-2021-051073 |
| [13] | Plaza-Manzano G, Gómez-Chiguano GF, Cleland JA, et al. (2020) Effectiveness of percutaneous electrical nerve stimulation for musculoskeletal pain: a systematic review and meta-analysis. Eur J Pain 24: 1023-1044. https://doi.org/10.1002/ejp.1559 |
| [14] | Mahran A, Baaklini G, Hassani D, et al. (2019) Sacral neuromodulation treating chronic pelvic pain: a meta-analysis and systematic review of the literature. Int Urogynecol J 30: 1023-1035. https://doi.org/10.1007/s00192-019-03898-w |
| [15] | Page MJ, McKenzie JE, Bossuyt PM, et al. (2021) The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. PLoS Med 18: e1003583. https://doi.org/10.1371/journal.pmed.1003583 |
| [16] | Downs SH, Black N (1998) The feasibility of creating a checklist for the assessment of the methodological quality both of randomised and non-randomised studies of health care interventions. J Epidemiol Community Health 52: 377-384. https://doi.org/10.1136/jech.52.6.377 |
| [17] | Higgins JPT, Altman DG, Gotzsche PC, et al. (2011) The Cochrane Collaboration's tool for assessing risk of bias in randomised trials. BMJ 343: d5928. https://doi.org/10.1136/bmj.d5928 |
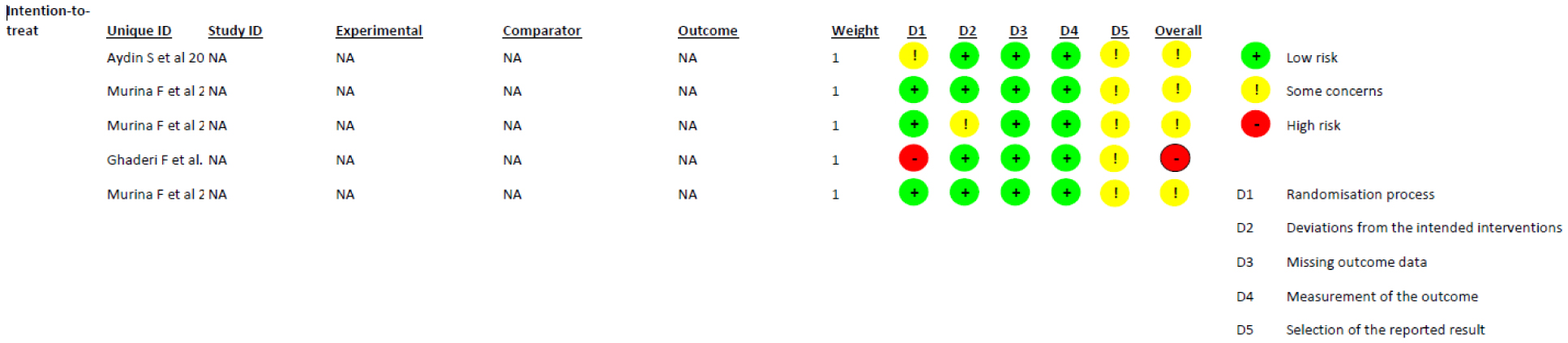
| [18] | Ghaderi F, Bastani P, Hajebrahimi S, et al. (2019) Pelvic floor rehabilitation in the treatment of women with dyspareunia: a randomized controlled clinical trial. Int Urogynecol J 30: 1849-1855. https://doi.org/10.1007/s00192-019-04019-3 |
| [19] | Murina F, Felice R, Di Francesco S, et al. (2018) Vaginal diazepam plus transcutaneous electrical nerve stimulation to treat vestibulodynia: a randomized controlled trial. Eur J Obstet Gynecol Reprod Biol 228: 148-153. https://doi.org/10.1016/j.ejogrb.2018.06.026 |
| [20] | Aydın S, Arıoğlu Aydın Ç, Batmaz G, et al. (2015) Effect of vaginal electrical stimulation on female sexual functions: a randomized study. J Sex Med 12: 463-469. https://doi.org/10.1111/jsm.12788 |
| [21] | Murina F, Graziottin A, Felice R, et al. (2013) Vestibulodynia: synergy between palmitoylethanolamide+ transpolydatin and transcutaneous electrical nerve stimulation. J Low Genit Tract Dis 17: 111-116. https://doi.org/10.1097/LGT.0b013e3182652316 |
| [22] | Murina F, Bianco V, Radici G, et al. (2008) Transcutaneous electrical nerve stimulation to treat vestibulodynia: a randomised controlled trial. BJOG 115: 1165-1170. https://doi.org/10.1111/j.1471-0528.2008.01803.x |
| [23] | Chalmers KJ, Madden VJ, Hutchinson MR, et al. (2016) Local and systemic inflammation in localized, provoked vestibulodynia: a systematic review. Obstet Gynecol 128: 337-347. https://doi.org/10.1097/AOG.0000000000001510 |
| [24] | Nappi RE, Cucinella L, Martella S, et al. (2016) Female sexual dysfunction (FSD): Prevalence and impact on quality of life (QoL). Maturitas 94: 87-91. https://doi.org/10.1016/j.maturitas.2016.09.013 |
| [25] | Laumann EO, Paik A, Rosen RC (1999) Sexual dysfunction in the United States: prevalence and predictors. JAMA 281: 537-544. https://doi.org/10.1001/jama.281.6.537 |
| [26] | Jaafarpour M, Khani A, Khajavikhan J, et al. (2013) Female sexual dysfunction: prevalence and risk factors. JCDR 7: 2877-2880. https://doi.org/10.7860/JCDR/2013/6813.3822 |
| [27] | Bergeron S, Corsini-Munt S, Aerts L, et al. (2015) Female sexual pain disorders: a review of the literature on etiology and treatment. Curr Sex Health Rep 7: 159-169. https://doi.org/10.1007/s11930-015-0053-y |
| [28] | Cottrell AM, Schneider MP, Goonewardene S, et al. (2020) Benefits and harms of electrical neuromodulation for chronic pelvic pain: a systematic review. Eur Urol Focus 6: 559-671. https://doi.org/10.1016/j.euf.2019.09.011 |
| [29] | Plaza-Manzano G, Gómez-Chiguano GF, Cleland JA, et al. (2020) Effectiveness of percutaneous electrical nerve stimulation for musculoskeletal pain: a systematic review and meta-analysis. Eur J Pain 24: 1023-1044. https://doi.org/10.1002/ejp.1559 |
| [30] | Ruiz BC, Outeiriño XP, Martı́nez PC, et al. (2004) Peripheral afferent nerve stimulation for treatment of lower urinary tract irritative symptoms. Eur Urol 45: 65-69. https://doi.org/10.1016/j.eururo.2003.08.012 |
| [31] | Beltran-Alacreu H, Serrano-Muñoz D, Martín-Caro Álvarez D, et al. (2022) Percutaneous versus transcutaneous electrical nerve stimulation for the treatment of musculoskeletal pain. A systematic review and meta-analysis. Pain Med 23: 1387-1400. https://doi.org/10.1093/pm/pnac027 |
| [32] | Melzack R, Wall PD (1967) Pain mechanisms: a new theory. Surv Anesthesiol 11: 89-90. https://doi.org/10.1097/00132586-196704000-00002 |
| [33] | Liebano RE, Rakel B, Vance CGT, et al. (2011) An investigation of the development of analgesic tolerance to TENS in humans. Pain 152: 335-342. https://doi.org/10.1016/j.pain.2010.10.040 |
| [34] | Plevnik S, Janez J (1979) Maximal electrical stimulation for urinary incontinence. Urology 14: 638-645. https://doi.org/10.1016/0090-4295(79)90545-4 |
Figures(2) / Tables(5)

López-López Laura, Villalobos-Santos Lourdes, Del Castillo-Matías Rocío, Torres-Sánchez Irene, Díaz-Mohedo Esther. Transcutaneous electrical nerve stimulation in the treatment of women with genito-pelvic pain penetration disorders: a systematic review[J]. AIMS Medical Science, 2024, 11(3): 348-360. doi: 10.3934/medsci.2024024







 DownLoad:
DownLoad: