Our investigation is motivated by the need to design bilayer membranes with tunable interfacial and mechanical properties for use in a range of applications, such as targeted drug delivery, sensing and imaging. We draw inspiration from biological cell membranes and focus on their principal constituents. In this paper, we present our results on the role of molecular architecture on the interfacial, structural and dynamical properties of bio-inspired membranes. We focus on four lipid architectures with variations in the head group shape and the hydrocarbon tail length. Each lipid species is composed of a hydrophilic head group and two hydrophobic tails. In addition, we study a model of the Cholesterol molecule to understand the interfacial properties of a bilayer membrane composed of rigid, single-tail molecular species. We demonstrate the properties of the bilayer membranes to be determined by the molecular architecture and rigidity of the constituent species. Finally, we demonstrate the formation of a stable mixed bilayer membrane composed of Cholesterol and one of the phospholipid species. Our approach can be adopted to design multi-component bilayer membranes with tunable interfacial and mechanical properties. We use a Molecular Dynamics-based mesoscopic simulation technique called Dissipative Particle Dynamics that resolves the molecular details of the components through soft-sphere coarse-grained models and reproduces the hydrodynamic behavior of the system over extended time scales.
Citation: Koufos Evan, Muralidharan Bharatram, Dutt Meenakshi. Computational Design of Multi-component Bio-Inspired Bilayer Membranes[J]. AIMS Materials Science, 2014, 1(2): 103-120. doi: 10.3934/matersci.2014.2.103
Our investigation is motivated by the need to design bilayer membranes with tunable interfacial and mechanical properties for use in a range of applications, such as targeted drug delivery, sensing and imaging. We draw inspiration from biological cell membranes and focus on their principal constituents. In this paper, we present our results on the role of molecular architecture on the interfacial, structural and dynamical properties of bio-inspired membranes. We focus on four lipid architectures with variations in the head group shape and the hydrocarbon tail length. Each lipid species is composed of a hydrophilic head group and two hydrophobic tails. In addition, we study a model of the Cholesterol molecule to understand the interfacial properties of a bilayer membrane composed of rigid, single-tail molecular species. We demonstrate the properties of the bilayer membranes to be determined by the molecular architecture and rigidity of the constituent species. Finally, we demonstrate the formation of a stable mixed bilayer membrane composed of Cholesterol and one of the phospholipid species. Our approach can be adopted to design multi-component bilayer membranes with tunable interfacial and mechanical properties. We use a Molecular Dynamics-based mesoscopic simulation technique called Dissipative Particle Dynamics that resolves the molecular details of the components through soft-sphere coarse-grained models and reproduces the hydrodynamic behavior of the system over extended time scales.

| [1] | Alexeev A, Uspal WE, Balazs AC (2008) Harnessing janus nanoparticles to create controllable pores in membranes.ACS Nano 2: 1117-1122. |
| [2] | Israelachvili JN (2010) Intermolecular and Surface Forces, 3 Eds..Waltham: Elsevier Science . |
| [3] | Almeida P, Vaz W (1995) Lateral diffusion in membranes.Handb Biol Phys 1: 305-357. |
| [4] | Monticelli L, Salonen E, Ke PC, et al. (2009) Effects of carbon nanoparticles on lipid membranes: a molecular simulation perspective.Soft Mat 5: 4433-4445. |
| [5] | Albanese A, Tang PS, Chan WCW (2012) The effect of nanoparticle size, shape, and surface chemistry on biological systems.Annu Rev Biomed Eng 14: 1-16. |
| [6] | Ding H, Ma Y (2012) Role of physicochemical properties of coating ligands in receptor-mediated endocytosis of nanoparticles.Biomaterials 33: 5798-5802. |
| [7] | Balme S, Janot J, Berado L, et al. (2011) New bioinspired membrane made of a biological ion channel confined into the cylindrical nanopore of a solid-state polymer.Nano Lett 11: 712-716. |
| [8] | Zhao L, Feng SS (2004) Effects of lipid chain length on molecular interactions between paclitaxel and phospholipid within model biomembranes.J Colloid Interface Sci 274: 55-68. |
| [9] | Baszkin A, Norde W (1999) Physical Chemistry of Biological Interfaces, 1 Eds..Boca Raton: CRC Press . |
| [10] | Hoogerbrugge PJ, Koelman J (1992) Simulating Microscopic Hydrodynamic Phenomena with Dissipative Particle Dynamics.Europhys Lett 19: 155-160. |
| [11] | Lin CM, Li CS, Sheng YJ, et al. (2012) Size-dependent properties of small unilamellar vesicles formed by model lipids.Langmuir 28: 689-700. |
| [12] | Schmid F, Schick M (1994) Monte Carlo study of interfacial properties in an amphiphilic system.Phys Rev E 49: 494-500. |
| [13] | Laradji M, Kumar P (2011) Advances in Planar Lipid Bilayers and Liposomes.In: Iglic A, 1 ed., Waltham: Elsevier Science . |
| [14] | Farago O (2008) Mode excitation Monte Carlo simulations of mesoscopically large membranes.J Chem Phys 128: 184105. |
| [15] | Klein ML, Shinoda W (2008) Large-scale molecular dynamics simulations of self-assembling systems.Science 321: 798-800. |
| [16] | Heller H, Schaefer M, Schulten K (1993) Molecular dynamics simulation of a bilayer of 200 lipids in the gel and in the liquid crystal phase.J Phys Chem 97: 8343-8360. |
| [17] | Wang Y, Jiang W, Yan T, et al. (2007) Understanding ionic liquids through atomistic and coarse-grained molecular dynamics simulations.Acc Chem Res 40: 1193-1199. |
| [18] | Cooke IR, Deserno M (2006) Coupling between lipid shape and membrane curvature.Biophys J 91: 487-495. |
| [19] | Brown FLH (2011) Continuum simulations of biomembrane dynamics and the importance of hydrodynamic effects.Q Rev Biophys 44: 391-432. |
| [20] | Aydin F, Dutt MIn Preparation. |
| [21] | Groot RD, Warren PB (1997) Dissipative particle dynamics: Bridging the gap between atomistic and mesoscopic simulation.J Chem Phys 107: 4423. |
| [22] | Gao L, Shillcock J, Lipowsky R (2007) Improved dissipative particle dynamics simulations of lipid bilayers.J Chem Phys 126: 01510. |
| [23] | Gullingsrud J, Schulten K (2004) Lipid bilayer pressure profiles and mechanosensitive channel gating.Biophys J 86: 3496-3509. |
| [24] | Dickson CJ, Rosso L, Betz RM, et al. (2012) GAFFlipid: a General Amber Force Field for the accurate molecular dynamics simulation of phospholipid.Soft Mat 8: 9617. |
| [25] | Dutt M, J. Nayhouse M, Kuksenok O, et al. (2011) Interactions of End-functionalized Nanotubes with Lipid Vesicles: Spontaneous Insertion and Nanotube Self-Organization.Curr Nanosci 7: 699-715. |
| [26] | Dutt M, Kuksenok O, Nayhouse MJ, et al. (2011) Modeling the self-assembly of lipids and nanotubes in solution: forming vesicles and bicelles with transmembrane nanotube channels.ACS Nano 5: 4769-4782. |
| [27] | Dutt M, Kuksenok O, Little SR, et al. (2011) Forming transmembrane channels using end-functionalized nanotubes.Nanoscale 3: 240-250. |
| [28] | Dutt M, Kuksenok O, Balazs AC (2012) Designing Tunable Bio-nanostructured Materials via Self-Assembly of Amphiphilic Lipids and Functionalized Nanotubes.MRS Proc 1464: mrss12-1464-rr08-09. |
| [29] | Dutt M, Kuksenok O, Balazs AC (2013) Nano-pipette directed transport of nanotube transmembrane channels and hybrid vesicles.MRS Proc 5: 9773-9784. |
| [30] | Frenkel D, Smit B (2002) Understnading Molecular Simulation: From Algorithms to Applications, 2 Eds..San Diego: Academic Press . |
| [31] | Alexeev A, Balazs AC (2007) Designing smart systems to selectively entrap and burst microcapsules.Soft Mat 3: 1500. |
| [32] | Orsi M, Essex JW (2013) Physical properties of mixed bilayers containing lamellar and nonlamellar lipids: insights from coarse-grain molecular dynamics simulations.Faraday Discuss 161: 249-272. |
| [33] | Schulz M, Olubummo A, Binder WH (2012) Beyond the lipid-bilayer: interaction of polymers and nanoparticles with membranes.Soft Mat 8: 4849-4864. |
| [34] | Lai K, Wang B, Zhang Y, et al. (2013) Computer simulation study of nanoparticle interaction with a lipid membrane under mechanical stress.Phys Chem Chem Phys 15: 270-278. |
| [35] | Deserno M (2009) Mesoscopic membrane physics: concepts, simulations, and selected applications.Macromol Rapid Commun 30: 752-757. |
| [36] | Bennun SV, Hoopes MI, Xing C, et al. (2009) Coarse-grained modeling of lipids.Chem Phys Lipids 159: 59-66. |
| [37] | Boal DH (2012) Mechanics of the Cell, 2 Eds..Cambridge: Cambridge University Press . |
| [38] | Zhong OY, Helfrich W (1987) Instability and Deformation of a Spherical Vesicle by Pressure.Phys Rev Lett 59: 2486-2488. |
| [39] | Lipowsky R (1991) The conformation of membranes.Nature 349: 475-478. |
| [40] | McIntosh TJ, Simon SA (2006) Roles of bilayer material properties in function and distribution of membrane proteins.Annu Rev Biophys Biomol Struct 35: 177-198. |
| [41] | Garcia ML (2004) Ion channels: gate expectations.Nature 430: 153-155. |
| [42] | Lin CM, Li CS, Sheng YJ, et al. (2012) Size-dependent properties of small unilamellar vesicles formed by model lipids.Langmuir 28: 689-700. |
| [43] | Chen H, Ruckinstein E (2011) Aggregation of nanoparticles in a block copolymer bilayer.J Coll Int Sci 363: 573-578. |
| [44] | Chen H, Ruckinstein E (2011) Nanoparticle aggregation in a block copolymer.J Chem Phys 131: 244904. |
| [45] | Venturoli M, Smit B, Sperotto MM (2005) Simulation studies of protein-induced bilayer deformations, and lipid-induced protein tilting, on a mesoscopic model for lipid bilayers with embedded proteins.Biophys J 88: 1778-1798. |
| [46] | de Meyer F, Smit B (2009) Effect of cholesterol on the structure of a phospholipid bilayer.Proc Natl Acad Sci 106: 3654-3658. |
| [47] | Koelman J, Hoogerbrugge PJ (1993) Dynamic Simulations of Hard-Sphere Suspensions Under Steady Shear.Europhys Lett 21: 363-368. |
| [48] | Venturoli M, Maddalena SM, Kranenburg M, et al. (2006) Mesoscopic models of biological membranes.Phys Rep 437: 1-54. |
| [49] | Koufos E, Dutt M (2013) Designing Nanostructured Hybrid Inorganic-biological Materials via the Self-assembly.MRS Proc 1569: mrss13-1569-ll02-03. |
| [50] | Kuksenok O, Balazs A (2007) Modeling multicomponent reactive membranes.Phys Rev E 75: 051906. |
| [51] | Zan GH, Tan C, Deserno M, et al. (2012) Hemifusion of giant unilamellar vesicles with planar hydrophobic surfaces: a fluorescence microscopy study.Soft Mat 8: 10877. |
| [52] | Weikl TR, Groves JT, Lipowsky R (2002) Pattern formation during adhesion of multicomponent membranes.Europhys Lett 59: 916-922. |
| [53] | Plimpton S (1995) Fast Parallel Algorithms for Short-Range Molecular Dynamics.J Comput Phys 117: 1-19. |
| [54] | Kučerka N, Nieh MP, Katsaras J (2011) Fluid phase lipid areas and bilayer thicknesses of commonly used phosphatidylcholines as a function of temperature.Biochim Biophys Acta 1808: 2761-2767. |
| [55] | Smith KA, Jasnow D, Balazs AC (2007) Designing synthetic vesicles that engulf nanoscopic particles.J Chem Phys 127: 084703. |
| [56] | Illya G, Lipowsky R, Shillcock JC (2005) Effect of chain length and asymmetry on material properties of bilayer membranes.J Chem Phys 122: 24490. |
| [57] | Goetz R, Lipowsky R (1998) Computer simulations of bilayer membranes: Self-assembly and interfacial tension.J Chem Phys 108: 7397-7409. |
| [58] | Cooke I, Kremer K, Deserno M (2005) Tunable generic model for fluid bilayer membranes.Phys Rev E 72: 011506. |
| [59] | Feller SE, Pastor RW (1999) Constant surface tension simulations of lipid bilayers: The sensitivity of surface areas and compressibilities.J Chem Phys 111: 1281-1287. |
| [60] | Smit B, Kranenburg M, Sperotto MM, et al. (2006) Computer Simulations in Condensed Matter: From Materials to Chemical Biology, Ferrario M, Ciccotti G, Binder K, 1 Eds..Berlin: Springer . |
| [61] | Turnbull D, Cohen MH (1970) On the Free-Volume Model of the Liquid-Glass Transition.J Chem Phys 52: 3038-3041. |
| [62] | Grest GS, Cohen MH (1981) Liquids, Glasses, and the Glass Transition: A Free-Volume Approach, Prigogine I, Rice SA.Adv Chem Phys 48: 455-525. |
| [63] | Almeida PFF, Vaz WLC, Thompson TE (1992) Lateral diffusion in the liquid phases of dimyristoylphosphatidylcholine/cholesterol lipid bilayers: a free volume analysis.Biochemistry 31: 6739-6747. |
| [64] | Turnbull D, Cohen MH (1961) Free-Volume Model of the Amorphous Phase: Glass Transition.J Chem Phys 34: 120-124. |
| [65] | Filippov A, Orädd Gäin model membranes studied by pulsed-field gradient-NMR: the role of lipid polyunsaturation.Biophys J 93: 3182-3190. |
| [66] | Bernardino de la Serna J, Orädd G, Bagatolli LA, et al. (2009) Segregated phases in pulmonary surfactant membranes do not show coexistence of lipid populations with differentiated dynamic properties.Biophys J 97: 1381-1389. |
| [67] | Falck E, Róg T, Karttunen M, et al. (2008) Lateral diffusion in lipid membranes through collective flows.J Am Chem Soc 130: 44-45. |
| [68] | Busch S, Smuda C, Pardo LC, et al. (2010) Molecular mechanism of long-range diffusion in phospholipid membranes studied by quasielastic neutron scattering.J Am Chem Soc 132: 3232-3233. |
| [69] | Apajalahti T, Niemela P, Govindan P, et al. (2010) Concerted diffusion of lipids in raft-like membranes.Faraday Discuss 144: 411-430. |
| [70] | Allen P, Tildesley DJ (1987) Computer Simulation of Liquids, 1 Eds..New York: Oxford University Press . |
| [71] | Vaz WLC, Clegg RM, Hallmann D (1985) Translational diffusion of lipids in liquid crystalline phase phosphatidylcholine multibilayers.A comparison of experiment with theory. Biochemistry 24: 781-786. |
| [72] | Egberts E, Berendsen HJC (1988) Molecular dynamics simulation of a smectic liquid crystal with atomic detail.J Chem Phys 89: 3718-3732. |
| [73] | Keffer D (2001) The Working Man's Guide to Obtaining Self Diffusion Coefficients from Molecular Dynamics Simulations.Available from: http://www.cs.unc.edu/Research/nbody/pubs/external/Keffer/selfD.pdf. . |
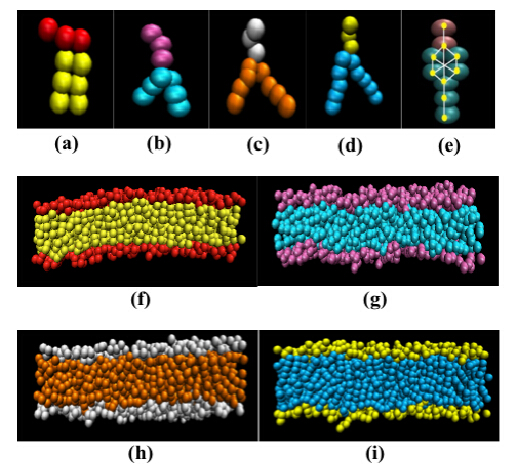
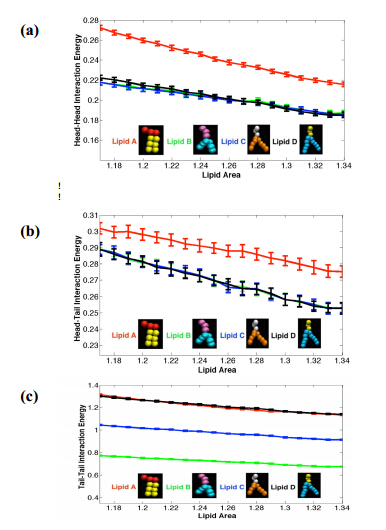
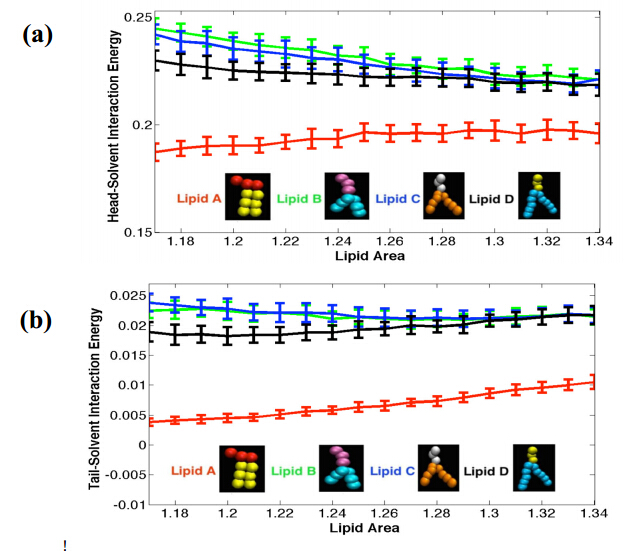

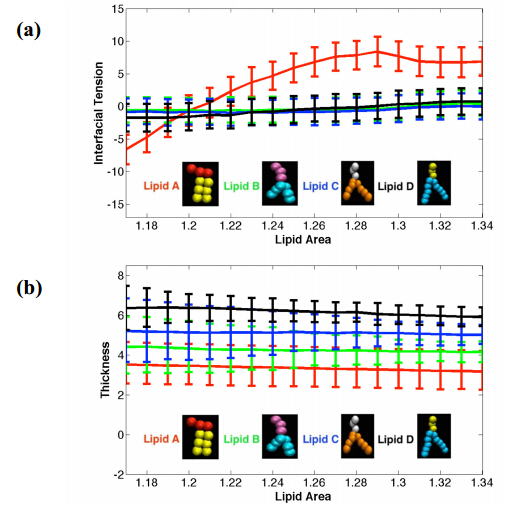
Figures(6)

Koufos Evan, Muralidharan Bharatram, Dutt Meenakshi. Computational Design of Multi-component Bio-Inspired Bilayer Membranes[J]. AIMS Materials Science, 2014, 1(2): 103-120. doi: 10.3934/matersci.2014.2.103







 DownLoad:
DownLoad: