Citation: Michael Sebastiano, Xiaolei Chu, Fikret Aydin, Leebyn Chong, Meenakshi Dutt. Interactions of Bio-Inspired Membranes with Peptides and Peptide-Mimetic Nanoparticles[J]. AIMS Materials Science, 2015, 2(3): 303-318. doi: 10.3934/matersci.2015.3.303

| [1] | Alberts B, Johnson A, Lewis J, et al. (2007) Molecular Biology of the Cell, Garland Science: New York. |
| [2] | Brannigan G, Brown FLH (2005) Composition Dependence of Bilayer Elasticity. J Chem Phys 122: 07490. |
| [3] | Shillcock JC, Lipowsky R (2002) Equilibrium structure and lateral stress distribution of amphiphilic bilayers from dissipative particle dynamics simulations. J Chem Phys 117: 5048-5061. |
| [4] | Lipowsky R, Sackmann E (1995) Structure and dynamics of membranes, Handbook of biological physics, Elsevier, Amsterdam. |
| [5] | Petelska AD, Figaszewski ZA (2002) Effect of pH on the Interfacial Tension of Lipid Bilayer Membrane. Biophys J 1561:135-146. |
| [6] |
Cooke IR, Kremer K, Deserno M (2005) Tunable Generic Model for Fluid Bilayer Membranes. Phys Rev E 72: 011506. doi: 10.1103/PhysRevE.72.011506

|
| [7] |
Laradji M, Kumar PBS (2004) Dynamics of Domain Growth in Self-assembled Fluid Vesicles. Phys Rev Lett 93: 198105. doi: 10.1103/PhysRevLett.93.198105
|
| [8] |
Laradji M, Kumar PBS (2005) Domain Growth, Budding, and Fission in Phase Separating Self-assembled Fluid Bilayers. J Chem Phys 123: 224902. doi: 10.1063/1.2102894
|
| [9] |
Ramachandran S, Laradji M, Kumar PBS (2009) Lateral Organization of Lipids in Multi-component Liposomes. J Phys Soc Jpn 78: 041006. doi: 10.1143/JPSJ.78.041006
|
| [10] |
Taniguchi T (1996) Shape Deformation and Phase Separation Dynamics of Two-component Vesicles. Phys Rev Lett 76: 4444-4447. doi: 10.1103/PhysRevLett.76.4444
|
| [11] | Fan J, Han T, Haataja M (2010) Hydrodynamic Effects on Spinodal Decomposition Kinetics in Planar Lipid Bilayer Membranes. J Chem Phys 133: 235101. |
| [12] |
Stanich CA, Honerkamp-Smith AR, Putzel GG, et al. (2013) Coarsening Dynamics of Domains in Lipid Membranes. Biophys J 105: 444-454. doi: 10.1016/j.bpj.2013.06.013
|
| [13] |
Veatch SL, Keller SL (2003) Separation of Liquid Phases in Giant Vesicles of Ternary Mixtures of Phospholipids and Cholesterol. Biophys J 85:3074-3083. doi: 10.1016/S0006-3495(03)74726-2
|
| [14] |
Esposito C, Tian A, Melamed S, et al. (2007) Flicker Spectroscopy of Thermal Lipid Bilayer Domain Boundary Fluctuations. Biophys J 93: 3169-3181. doi: 10.1529/biophysj.107.111922
|
| [15] | Lipowsky R (1992) Budding of Membranes Induced by Intramembrane Domains. J Phys II 2: 1825. |
| [16] |
Bagatolli LA, Gratton E (2001) Direct Observation of Lipid Domains in Free Standing Bilayers Using Two-photon Excitation Fluorescence Microscopy. J Fluorescence 11: 141-160. doi: 10.1023/A:1012228631693
|
| [17] |
Ramachandran S, Komura S, Gommper G (2010) Effects of an Embedding Bulk Fluid on Phase Separation Dynamics in a Thin Liquid Film. EPL 89: 56001. doi: 10.1209/0295-5075/89/56001
|
| [18] |
Ursell TS, Klug WS, Phillips R (2009) Morphology and Interaction between Lipid Domains. Proc Natl Acad Sci U S A 106: 13301. doi: 10.1073/pnas.0903825106
|
| [19] |
Bagatolli L, Kumar PBS (2009) Phase Behavior of Multicomponent Membranes: Experimental and Compuatational Techniques. Soft Matter 5: 3234-3248. doi: 10.1039/b901866b
|
| [20] | Marrink SJ, de Vries AH, Tieleman DP (2009) Lipids on the Move: Simulations of Membrane Pores, Domains, Stalks and Curves. Biochim Biophys Acta Biomembr 1788: 149-168. |
| [21] |
Lipowsky R (2002) Domains and Rafts in Membranes—Hidden Dimensions of Selforganization. J Biol Phys 28: 195-210. doi: 10.1023/A:1019994628793
|
| [22] | Simons K, Vaz WLC (2004) Model Systems, Lipid Rafts, and Cell Membranes. Annu Rev Biophys Biomol Struct 3: 269. |
| [23] |
Barberousse A, Franceschelli S, Imbert C (2009) Computer Simulations as Experiments. Synthese 169: 557-574. doi: 10.1007/s11229-008-9430-7
|
| [24] | Farago O (2003) “Water-free” Computer Model for Fluid Bilayer Membranes. O J Chem Phys 119: 596-605. |
| [25] |
Brannigan G, Brown FLH (2004) Solvent-free Simulations of Fluid Membrane Bilayers. J Chem Phys 120: 1059. doi: 10.1063/1.1625913
|
| [26] | Shillcock JC (2012) Spontaneous Vesicle Self-Assembly: A Mesoscopic View of Membrane Dynamics. Langmuir 28: 541-547. |
| [27] | Tieleman DP, Leontiadau H, Mark AE, et al. (2003) Simulation of Pore Formation in Lipid Bilayers by Mechanical Stress and Electric Fields. J Am Chem Soc125: 6382-6383. |
| [28] |
Damodaran KV, Merz KM (1994) A Comparison of DMPC- and DLPE-based Lipid Bilayers. Biophys J 66: 1076-1087. doi: 10.1016/S0006-3495(94)80889-6
|
| [29] |
Moore PB, Lopez CF, Klein ML (2001) Dynamical Properties of a Hydrated Lipid Bilayer from a Multinanosecond Molecular Dynamics Simulation. Biophys J 81: 2484-2494. doi: 10.1016/S0006-3495(01)75894-8
|
| [30] |
Essmann U, Perera L, Berkowitz ML (1995) The Origin of the Hydration Interaction of Lipid Bilayers from MD Simulation of Dipalmitoylphosphatidylcholine Membranes in Gel and Liquid Crystalline Phases. Langmuir 11: 4519-4531. doi: 10.1021/la00011a056
|
| [31] |
Cooke IR, Deserno M (2005) Solvent-free Model for Self-assembling Fluid Bilayer Membranes: Stabilization of the Fluid Phase based on Broad Attractive Tail Potentials. J Chem Phys 123: 224710. doi: 10.1063/1.2135785
|
| [32] |
West B, Schmid F (2010) Fluctuations and Elastic Properties of Lipid Membranes in the Gel L-beta State: A Coarse-grained Monte Carlo Study. Soft Matter 6: 1275. doi: 10.1039/b920978f
|
| [33] |
Farago O (2008) Mode Excitation Monte Carlo Simulations of Mesoscopically Large Membranes. J Chem Phys 128: 184105. doi: 10.1063/1.2918736
|
| [34] |
Farago O (2010) Fluctuation-induced Attraction between Adhesion Sites of Supported Membranes. Phys Rev E 81: 050902. doi: 10.1103/PhysRevE.81.050902
|
| [35] |
Farago O (2008) Membrane Fluctuations near a Plane Rigid Surface. Phys Rev E 78:051919. doi: 10.1103/PhysRevE.78.051919
|
| [36] | Dutt M, Nayhouse MJ, Kuksenok O, et al. (2011) Interactions of End-Functionalized Nanotubes with Lipid Vesicles: Spontaneous Insertion and Nanotube Self-organization. Current Nanoscience 7: 699-715. |
| [37] |
Dutt M, Kuksenok O, Nayhouse MJ, et al. (2011) Modeling the Self-Assembly of Lipids and Nanotubes in Solution: Forming Vesicles and Bicelles with Transmembrane Nanotube Channels. ACS Nano 5: 4769-4782. doi: 10.1021/nn201260r
|
| [38] |
Dutt M, Kuksenok O, Little SR, et al. (2011) Forming Transmembrane Channels Using End-Functionalized Nanotubes. Nanoscale. 3: 240-250. doi: 10.1039/C0NR00578A
|
| [39] | Dutt M, Kuksenok O, Little SR, et al. (2012) Designing Tunable Bio-nanostructured Materials via Self-assembly of Amphiphilic Lipids and Functionalized Nanotubes. MRS Spring 2012 Conference Proceedings; 1464. |
| [40] | Ludford P, Aydin F, Dutt M (2013) Design and Characterization of Nanostructured Biomaterials via the Self-assembly of Lipids. MRS Fall 2013 Conference Proceedings; 1498. |
| [41] | Koufos E, Dutt M (2013) Design of Nanostructured Hybrid Inorganic-biological Materials via Self-assembly. MRS Spring 2013 Conference Proceedings; 1569. |
| [42] |
Smith KA, Jasnow D, Balazs AC (2007) Designing Synthetic Vesicles that Engulf Nanoscopic Particles. J Chem Phys 127: 084703. doi: 10.1063/1.2766953
|
| [43] | Goetz R, Lipowsky R (1998) Computer Simulations of Bilayer Membranes: Self-assembly and Interfacial Tension. J Chem Phys 108: 7397-7409. |
| [44] | Kranenburg M, Venturoli M, Smit B. (2003) Phase Behavior and Induced Interdigitation in Bilayers Studied with Dissipative Particle Dynamics. J Phys Chem 41: 11491. |
| [45] |
Kranenburg M, Laforge C, Smit B (2004) Mesoscopic Simulations of Phase Transitions in Lipid Bilayers. Phys Chem Chem Phys 6: 4531-4534. doi: 10.1039/b410914g
|
| [46] | Yamamoto S, Maruyama Y, Hyodo S (2002) Dissipative Particle Dynamics Study of Spontaneous Vesicle Formation of Amphiphilic Molecules. J Chem Phys 116: 5842. |
| [47] |
Yamamoto S, Hyodo S (2003) Budding and Fission Dynamics of Two-Component Vesicles. J Chem Phys 118: 7937-7943. doi: 10.1063/1.1563613
|
| [48] |
Stevens MJ, Hoh JH, Woolf TB (2003) Insights into the Molecular Mechanism of Membrane Fusion from Simulation: Evidence for the Association of Splayed Tails. Phys Rev Lett 91: 188102. doi: 10.1103/PhysRevLett.91.188102
|
| [49] | Stevens MJ (2004) Coarse-grained Simulations of Lipid Bilayers. Chem Phys 121: 11942-11948. |
| [50] | Arkhipov A, Yin Y, Schulten K (2009) Membrane-bending Mechanism of Amphiphysin N-BAR Domains. Biophys J 97: 2727-2735. |
| [51] |
Shih AY, Arkhipov A, Freddolino PL, et al. (2006) A Coarse-grained Protein-lipid Model with Application to Lipoprotein Particles. J Phys Chem 110: 3674-3684. doi: 10.1021/jp0550816
|
| [52] |
Marrink SJ, Risselada HJ, Yefimov S, et al. (2007) The MARTINI Forcefield: Coarse-grained Model for Biomolecular Simulations. J Phys Chem B 111: 7812-7824. doi: 10.1021/jp071097f
|
| [53] | Wang Z, Frenkel DJ (2005) Modeling Flexible Amphiphilic Bilayers: A Solvent-free Off-lattice Monte Carlo Study. Chem Phys 122: 234711. |
| [54] |
Brannigan G, Philips PF, Brown FLH (2005) Flexible Lipid Bilayers in Implicit Solvent. Phys Rev E 72: 011915. doi: 10.1103/PhysRevE.72.011915
|
| [55] |
Noguchi H, Takasu M (2001) Self-assembly of Amphiphiles into Vesicles: A Brownian Dynamics Simulation. Phys Rev E 64: 041913. doi: 10.1103/PhysRevE.64.041913
|
| [56] |
Noguchi H (2002) Fusion and Toroidal Formation of Vesicles by Mechanical Forces: A Brownian Dynamics Simulation. J Chem Phys 117: 8130-8137. doi: 10.1063/1.1510114
|
| [57] |
Katsov K, Mueller M, Schick M (2004) Field Theoretic Study of Bilayer Membrane Fusion I Hemifusion Mechanism. Biophys J 87: 3277. doi: 10.1529/biophysj.103.038943
|
| [58] | Schick M (2012) Membranes: A Field-theoretic Description. Encyclopedia of Biophysics. Roberts, G.C.K., Ed., Springer-Verlag: Berlin Heidelberg. |
| [59] |
May S, Kozlovsky Y, Ben-Shaul A, et al. (2004) Tilt Modulus of a Lipid Monolayer. Eur Phys J E 14: 299-308. doi: 10.1140/epje/i2004-10019-y
|
| [60] |
May S (2000) A Molecular Model for the Line Tension of Lipid Membranes. Eur Phys J E 3: 37-44. doi: 10.1007/s101890070039
|
| [61] |
Lee WB, Mezzenga R, Fredrickson GH (2008) Self-consistent Field Theory for Lipid-based Liquid Crystals: Hydrogen Bonding Effect. J Chem Phys 128: 074504-074510. doi: 10.1063/1.2838624
|
| [62] | Ginzburg VV, Balijepalli S (2007) Modelling the Thermodynamics of the Interaction of Nanoparticles with Cell Membranes. Nano Lett 7: 3716-3722. |
| [63] |
Ayton G, Voth GA (2002) Bridging Microscopic and Mesoscopic Simulations of Lipid Bilayers. Biophys J 83: 3357-3370. doi: 10.1016/S0006-3495(02)75336-8
|
| [64] |
Wang ZJ, Deserno MA (2010) Systematically Coarse-grained Solvent-free Model for Quantitative Phospholipid Bilayer Simulations. J Phys Chem B 114: 11207. doi: 10.1021/jp102543j
|
| [65] |
Wang ZJ, Deserno M (2010) Systematic Implicit Solvent Coarse-graining of Bilayer Membranes: Lipid and Phase Transferability of the Force Field. New J Phys 12: 095004. doi: 10.1088/1367-2630/12/9/095004
|
| [66] |
Ge Z, Li Q, Wang Y (2014) Free energy Calculation of Nanodiamond-Membrane Association—The Effect of Shape and Surface Functionalization. J Chem Theory Comput 10: 2751-2758. doi: 10.1021/ct500194s
|
| [67] | Reid CVL, Ricci M, Silva PHJ, et al. (2014) Lipid tail protrusions mediate the insertion of nanoparticles into model cell membranes. Nat Commun 5: 4482. |
| [68] |
Wong-Ekkabut J, Baoukina S, Triampo W, et al. (2008) Computer simulation study of fullerene translocation through lipid membranes. Nat Nanotechnol 3: 363-368. doi: 10.1038/nnano.2008.130
|
| [69] |
Li Y, Chen X, Gu N (2008) Computational Investigation of Interaction between Nanoparticles and Membranes: Hydrophobic/Hydrophilic Effect. J Phys Chem B 112: 16647-16653. doi: 10.1021/jp8051906
|
| [70] |
Huang C, Zhang Y, Yuan H, et al. (2013) Role of Nanoparticle Geometry in Endocytosis: Laying Down to Stand Up. Nano Lett 13: 4546-4550. doi: 10.1021/nl402628n
|
| [71] |
Shi X, Bussche AVD, Hurt RH, et al. (2011) Cell entry of one-dimensional nanomaterials occurs by tip recognition and rotation. Nat Nanotechnol 6: 714-719. doi: 10.1038/nnano.2011.151
|
| [72] |
Illya G, Lipowsky R, Shillcock JC (2006) Two-component membrane material properties and domain formation from dissipative particle dynamics. J Chem Phys 125: 114710. doi: 10.1063/1.2353114
|
| [73] |
Groot RD, Warren PB (1997) Dissipative Particle Dynamics: Bridging the gap between atomistic and mesoscopic simulation. J Chem Phys 107: 4423-4435. doi: 10.1063/1.474784
|
| [74] |
Chou H, Tsao HK, Sheng YJ (2006) Morphologies of multicompartment micelles formed by triblock copolymers. J Chem Phys 125: 194903. doi: 10.1063/1.2390716
|
| [75] |
Ortiz V, Nielsen SO, Discher DE, et al. (2005) Disipative Particle Dyanmics simulations of polymerosome. J Phys Chem B 109: 17708-17714. doi: 10.1021/jp0512762
|
| [76] | Boek ES, Coveney PV, Lekkerkerker HNW, et al. (1997) Simulating rheology of dense colloidal suspensions using dissipative particle dynamics. Phys Rev E 55: 3124-3131. |
| [77] | Spenley NA (200) Scaling laws for polymers in dissipative particle dynamics, Europhys Lett 49: 534-540. |
| [78] |
Fan XJ, Phan-Thien N, Chen S, et al. (2006) Simulating flow of DNA suspension using dissipative particle dynamics. Phys Fluids 18: 063102. doi: 10.1063/1.2206595
|
| [79] |
Chem S, Phan-Thien N, Fan XJ, et al. (2004) Dissipative particle dynamics of polymer drops in periodic shear flow. J Non-Newtonian Fluid Mech 118: 65-81. doi: 10.1016/j.jnnfm.2004.02.005
|
| [80] |
Arai N, Yasuoka K, Zeng XC (2013) A vesicle cell under collision with a Janus or homogeneous nanoparticle: translocation dynamics and late-stage morphology. Nanoscale 5: 9089-9100. doi: 10.1039/c3nr02024j
|
| [81] |
Yang K, Ma Y (2010) Computer simulation of the translocation of nanoparticles with different shapes across a lipid bilayer. Nat Nanotechnol 5: 579-583. doi: 10.1038/nnano.2010.141
|
| [82] |
Ding H, Tian W, Ma Y (2012) Designing Nanoparticle Translocation through Membranes by Computer Simulations. ACS Nano 6: 1230-1238. doi: 10.1021/nn2038862
|
| [83] |
Chen X, Tian F, Zhang X, et al. (2013) Internalization pathways of nanoparticles and their interaction with a vesicle. Soft Matter 9: 7592-7600. doi: 10.1039/c3sm50931a
|
| [84] |
Arnarez C, Uusitalo JJ, Masman MF, et al. (2015) Dry Martini, a Coarse-Grained Force Field for Lipid Membrane Simulations with Implicit Solvent. J Chem Theory Comput 11: 260-275. doi: 10.1021/ct500477k
|
| [85] | Hall BA, Chetwynd AP, Sansom MSP (2011) Exploring Peptide-Membrane Interactions with Coarse-Grained MD Simulations. Biophys J 100: 1940-1948. |
| [86] |
Gkeka P, Sarkisov L (2010). Interactions of Phospholipid Bilayers with Several Classes of Amphiphilic α-Helical Peptides: Insights from Coarse-Grained Molecular Dynamics Simulations. J Phys Chem B 114: 826-839. doi: 10.1021/jp908320b
|
| [87] | Allen MP, Tildesley DJ (2001) Computer simulations of liquids, Clarendon Press, Oxford. |
| [88] |
Koufos E, Muralidharan B, Dutt M (2014) Computational Design of Multi-component Bio-Inspired Bilayer. AIMS Materials Science 1: 103-120. doi: 10.3934/matersci.2014.2.103
|
| [89] |
Milletti F (2012) Cell-Penetrating Peptides: Classes, Origin, and Current Landscape. Drug Discov Today 17: 850-860. doi: 10.1016/j.drudis.2012.03.002
|
| [90] |
Aydin F, Ludford P, Dutt M (2014) Phase segregation in bio-inspired multi-component vesicles encompassing double tail phospholipid species. Soft Matter 10: 6096-6108. doi: 10.1039/C4SM00998C
|
| [91] |
Aydin F, Uppaladadium G, Dutt M (2015) The Design of Shape-Tunable Hairy Vesicles. Colloids Surf B Biointerfaces 128: 268-275. doi: 10.1016/j.colsurfb.2015.01.049
|
| [92] |
Monticelli L, Kandasamy SK, Periole X, et al. (2008) The MARTINI Coarse-Grained Force Field: Extension to Proteins. J Chem Theory Comput 4: 819-834. doi: 10.1021/ct700324x
|
| [93] |
Feller SE, Pastor RW (1999) Constant surface tension simulations of lipid bilayers: The sensitivity of surface areas and compressibilities. J Chem Phys 111: 1281-1287. doi: 10.1063/1.479313
|
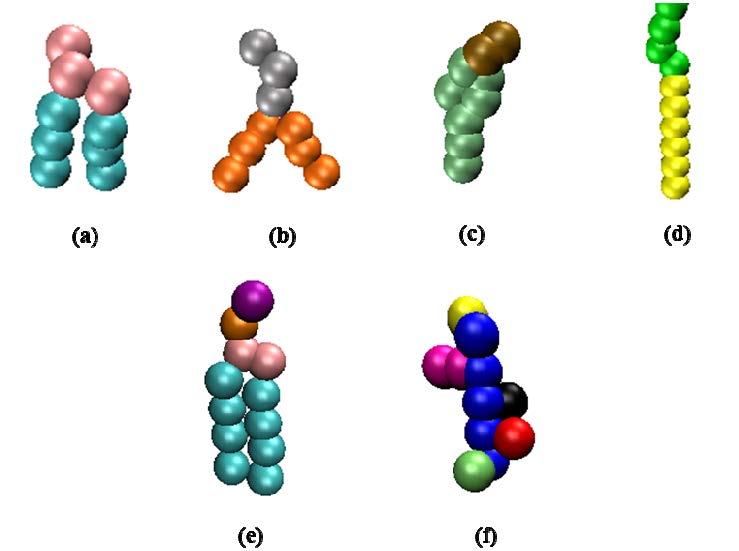
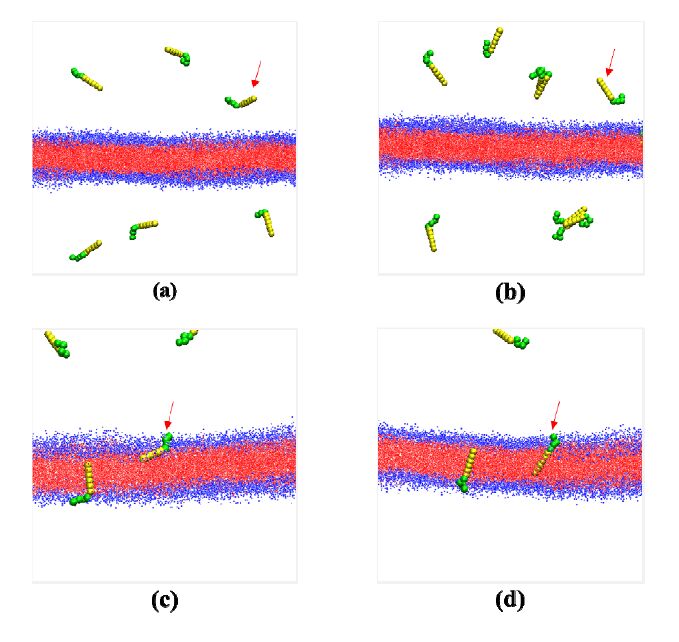
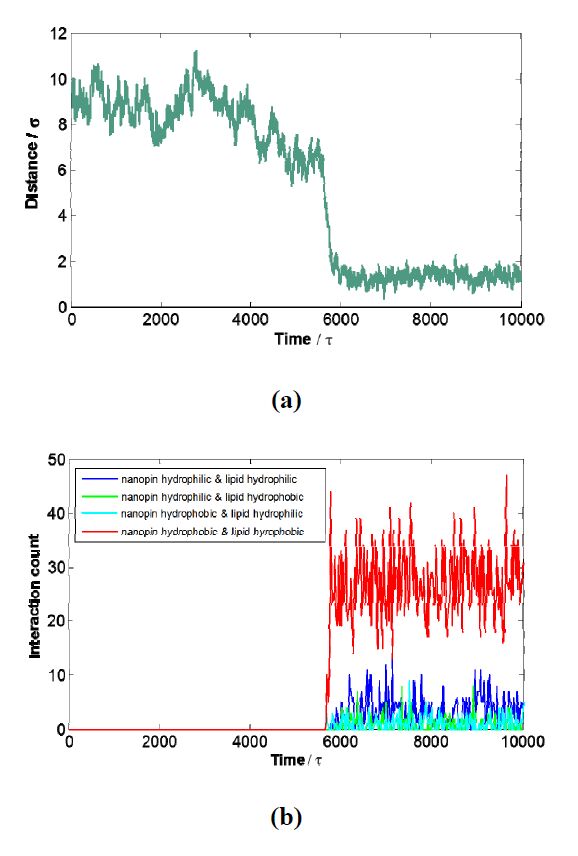
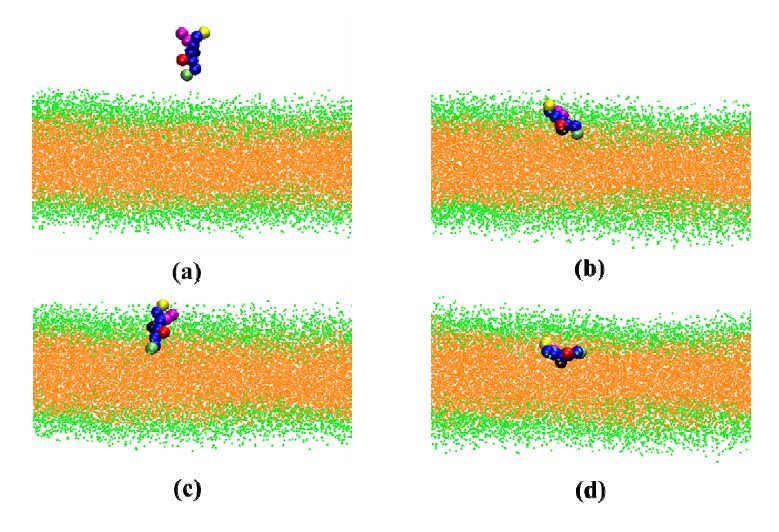
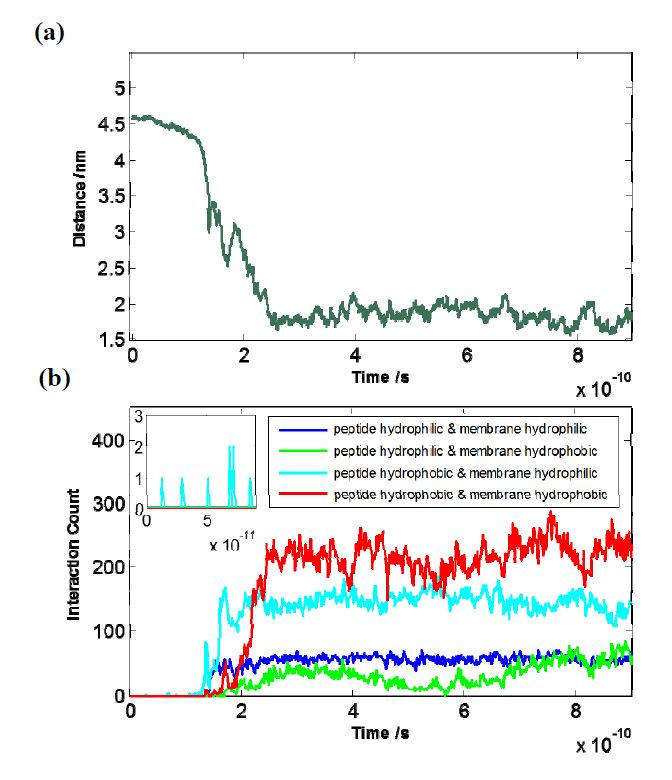
Figures(5)

Michael Sebastiano, Xiaolei Chu, Fikret Aydin, Leebyn Chong, Meenakshi Dutt. Interactions of Bio-Inspired Membranes with Peptides and Peptide-Mimetic Nanoparticles[J]. AIMS Materials Science, 2015, 2(3): 303-318. doi: 10.3934/matersci.2015.3.303







 DownLoad:
DownLoad: