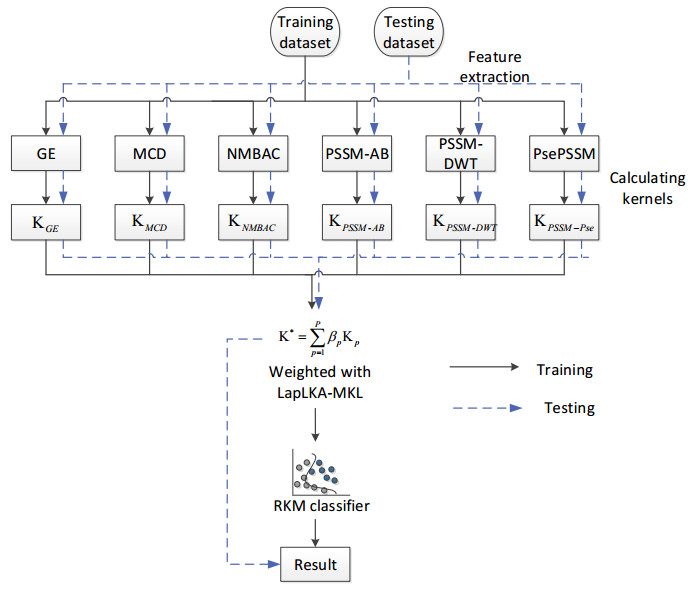
DNA-binding proteins (DBPs) play a critical role in the development of drugs for treating genetic diseases and in DNA biology research. It is essential for predicting DNA-binding proteins more accurately and efficiently. In this paper, a Laplacian Local Kernel Alignment-based Restricted Kernel Machine (LapLKA-RKM) is proposed to predict DBPs. In detail, we first extract features from the protein sequence using six methods. Second, the Radial Basis Function (RBF) kernel function is utilized to construct pre-defined kernel metrics. Then, these metrics are combined linearly by weights calculated by LapLKA. Finally, the fused kernel is input to RKM for training and prediction. Independent tests and leave-one-out cross-validation were used to validate the performance of our method on a small dataset and two large datasets. Importantly, we built an online platform to represent our model, which is now freely accessible via http://8.130.69.121:8082/.
Citation: Yuqing Qian, Tingting Shang, Fei Guo, Chunliang Wang, Zhiming Cui, Yijie Ding, Hongjie Wu. Identification of DNA-binding protein based multiple kernel model[J]. Mathematical Biosciences and Engineering, 2023, 20(7): 13149-13170. doi: 10.3934/mbe.2023586
DNA-binding proteins (DBPs) play a critical role in the development of drugs for treating genetic diseases and in DNA biology research. It is essential for predicting DNA-binding proteins more accurately and efficiently. In this paper, a Laplacian Local Kernel Alignment-based Restricted Kernel Machine (LapLKA-RKM) is proposed to predict DBPs. In detail, we first extract features from the protein sequence using six methods. Second, the Radial Basis Function (RBF) kernel function is utilized to construct pre-defined kernel metrics. Then, these metrics are combined linearly by weights calculated by LapLKA. Finally, the fused kernel is input to RKM for training and prediction. Independent tests and leave-one-out cross-validation were used to validate the performance of our method on a small dataset and two large datasets. Importantly, we built an online platform to represent our model, which is now freely accessible via http://8.130.69.121:8082/.

| [1] | M. J. Buck, J. D. Lieb, ChIP-chip: considerations for the design, analysis, and application of genome-wide chromatin immunoprecipitation experiments, Genomics, 83 (2004), 349–360. https://doi.org/10.1016/j.ygeno.2003.11.004 |
| [2] |
F. Cui, S. Li, Z. Zhang, M. Sui, C. Cao, A. E. Hesham, et al., DeepMC-iNABP: Deep learning for multiclass identification and classification of nucleic acid-binding proteins, Comput. Struct. Biotechnol. J., 20 (2022), 2020–2028. https://doi.org/10.1016/j.csbj.2022.04.029 doi: 10.1016/j.csbj.2022.04.029

|
| [3] |
F. Cajone, M. Salina, A. Benelli-Zazzera, 4-Hydroxynonenal induces a DNA-binding protein similar to the heat-shock factor, Biochem. J., 262 (1989), 977–979. https://doi.org/10.1042/bj2620977 doi: 10.1042/bj2620977
|
| [4] |
M. Gao, S. Jeffrey, DBD-Hunter: a knowledge-based method for the prediction of DNA-protein interactions, Nucleic Acids Res., 36 (2008), 3978–3992. https://doi.org/10.1093/nar/gkn332 doi: 10.1093/nar/gkn332
|
| [5] |
Y. Fang, Y. Guo, Y. Feng, M. Li, Predicting DNA-binding proteins: approached from Chou's pseudo amino acid composition and other specific sequence features, Amino Acids, 34 (2008), 103–109. https://doi.org/10.1007/s00726-007-0568-2 doi: 10.1007/s00726-007-0568-2
|
| [6] |
C. Cao, L. Mak, G. Jin, P. Gordon, K. Ye, Q. Long, PRESM: personalized reference editor for somatic mutation discovery in cancer genomics, Bioinformatics, 35 (2019), 1445–1452. https://doi.org/10.1093/bioinformatics/bty812 doi: 10.1093/bioinformatics/bty812
|
| [7] |
C. Cao, M. Greenberg, Q. Long, WgLink: reconstructing whole-genome viral haplotypes using L0+ L1-regularization, Bioinformatics, 37 (2021), 2744–2746. https://doi.org/10.1093/bioinformatics/btab076 doi: 10.1093/bioinformatics/btab076
|
| [8] |
C. Cao, J. He, L. Mak, D. Perera, D. Kwok, J. Wang, et al., Reconstruction of microbial haplotypes by integration of statistical and physical linkage in scaffolding, Mol. Biol. Evol., 38 (2021), 2660–2672. https://doi.org/10.1093/molbev/msab037 doi: 10.1093/molbev/msab037
|
| [9] |
Z. Zhang, F. Cui, W. Su, L. Dou, A. Xu, C. Cao, et al., webSCST: an interactive web application for single-cell RNA-sequencing data and spatial transcriptomic data integration, Bioinformatics, 38 (2022), 3488–3489. https://doi.org/10.1093/bioinformatics/btac350 doi: 10.1093/bioinformatics/btac350
|
| [10] |
Z. Zhang, F. Cui, C. Wang, L. Zhao, Q. Zou, Goals and approaches for each processing step for single-cell RNA sequencing data, Briefing Bioinf., 22 (2021), bbaa314. https://doi.org/10.1093/bib/bbaa314 doi: 10.1093/bib/bbaa314
|
| [11] |
F. Cui, Z. Zhang, C. Cao, Q. Zou, D. Chen, X. Su, Protein–DNA/RNA interactions: Machine intelligence tools and approaches in the era of artificial intelligence and big data, Proteomics, 22 (2022), 2100197. https://doi.org/10.1002/pmic.202100197 doi: 10.1002/pmic.202100197
|
| [12] |
B. Liu, J. Xu, S. Fan, R. Xu, J. Zhou, X. Wang, PseDNA‐Pro: DNA‐binding protein identification by combining Chou's PseAAC and physicochemical distance transformation, Mol. Inf., 34 (2015), 8–17. https://doi.org/10.1002/minf.201400025 doi: 10.1002/minf.201400025
|
| [13] |
B. Liu, S. Wang, X. Wang, DNA binding protein identification by combining pseudo amino acid composition and profile-based protein representation, Sci. Rep., 5 (2015), 15479. https://doi.org/10.1038/srep15479 doi: 10.1038/srep15479
|
| [14] |
Y. Ding, J. Tang, F. Guo, Identification of protein-ligand binding sites by sequence information and ensemble classifier, J. Chem. Inf. Model., 57 (2017), 3149–3161. https://doi.org/10.1021/acs.jcim.7b00307 doi: 10.1021/acs.jcim.7b00307
|
| [15] |
Y. Ding, J. Tang, F. Guo, Human protein subcellular localization identification via fuzzy model on Kernelized Neighborhood Representation, Appl. Soft Comput., 96 (2020), 106596. https://doi.org/10.1016/j.asoc.2020.106596 doi: 10.1016/j.asoc.2020.106596
|
| [16] |
G. Nimrod, M. Schushan, A. Szilágyi, C. Leslie, N. Ben-Tal, iDBPs: a web server for the identification of DNA binding proteins, Bioinformatics, 26 (2010), 692–693. https://doi.org/10.1093/bioinformatics/btq019 doi: 10.1093/bioinformatics/btq019
|
| [17] |
M. S. Rahman, S. Shatabda, S. Saha, M. Kaykobad, M. S. Rahman, DPP-PseAAC: a DNA-binding protein prediction model using Chou's general PseAAC, J. Theor. Biol., 452 (2018), 22–34. https://doi.org/10.1016/j.jtbi.2018.05.006 doi: 10.1016/j.jtbi.2018.05.006
|
| [18] |
K. C. Chou, Some remarks on protein attribute prediction and pseudo amino acid composition, J. Theor. Biol., 273 (2011), 236–247. https://doi.org/10.1016/j.jtbi.2010.12.024 doi: 10.1016/j.jtbi.2010.12.024
|
| [19] |
C. Cortes, V. Vapnik, Support-vector networks, Mach. Learn., 20 (1995), 273–297. https://doi.org/10.1007/BF00994018 doi: 10.1007/BF00994018
|
| [20] |
S. F. Altschul, T. L. Madden, A. A. Schäffer, J. Zhang, Z. Zhang, W. Miller, et al., Gapped BLAST and PSI-BLAST: a new generation of protein database search programs, Nucleic Acids Res., 25 (1997), 3389–3402. https://doi.org/10.1093/nar/25.17.3389 doi: 10.1093/nar/25.17.3389
|
| [21] |
Y. Ding, J. Tang, F. Guo, Identification of protein–ligand binding sites by sequence information and ensemble classifier, J. Chem. Inf. Model., 57 (2017), 3149–3161. https://doi.org/10.1021/acs.jcim.7b00307 doi: 10.1021/acs.jcim.7b00307
|
| [22] |
F. Guo, Y. Ding, Z. Li, J. Tang, Identification of protein-protein interactions by detecting correlated mutation at the interface, J. Chem. Inf. Model., 55 (2015), 2042–2049. https://doi.org/10.1021/acs.jcim.5b00320 doi: 10.1021/acs.jcim.5b00320
|
| [23] |
Y. Ding, J. Tang, F. Guo, Protein crystallization identification via fuzzy model on linear neighborhood representation, IEEE/ACM Trans. Comput. Biol. Bioinf., 18 (2019), 1986–1995. https://doi.org/10.1109/TCBB.2019.2954826 doi: 10.1109/TCBB.2019.2954826
|
| [24] |
M. Wang, J. Yang, G. Liu, Z. Xu, K. Chou, Weighted-support vector machines for predicting membrane protein types based on pseudo-amino acid composition, Protein Eng. Des. Sel., 17 (2004), 509–516. https://doi.org/10.1093/protein/gzh061 doi: 10.1093/protein/gzh061
|
| [25] |
M. Hayat, A. Khan, Predicting membrane protein types by fusing composite protein sequence features into pseudo amino acid composition, J. Theor. Biol., 271 (2010), 10–17. https://doi.org/10.1016/j.jtbi.2010.11.017 doi: 10.1016/j.jtbi.2010.11.017
|
| [26] |
Y. Qian, L. Jiang, Y. Ding, J. Tang, F. Guo, A sequence-based multiple kernel model for identifying DNA-binding proteins, BMC Bioinf., 22 (2021), 1–18. https://doi.org/10.1186/s12859-020-03875-x doi: 10.1186/s12859-020-03875-x
|
| [27] |
Y. Qian, H. Meng, W. Lu, Z. Liao, Y. Ding, H. Wu, Identification of DNA-binding proteins via hypergraph based laplacian support vector machine, Curr. Bioinf., 17 (2022), 108–117. https://doi.org/10.2174/1574893616666210806091922 doi: 10.2174/1574893616666210806091922
|
| [28] |
S. Zhao, Y. Ding, X. Liu, X. Su, HKAM-MKM: a hybrid kernel alignment maximization-based multiple kernel model for identifying DNA-binding proteins, Comput. Biol. Med., 145 (2022), 105395. https://doi.org/10.1016/j.compbiomed.2022.105395 doi: 10.1016/j.compbiomed.2022.105395
|
| [29] |
M. Sun, P. Tiwari, Y. Qian, Y. Ding, Q. Zou, MLapSVM-LBS: Predicting DNA-binding proteins via a multiple Laplacian regularized support vector machine with local behavior similarity, Knowledge-Based Syst., 250 (2022), 109174. https://doi.org/10.1016/j.knosys.2022.109174 doi: 10.1016/j.knosys.2022.109174
|
| [30] |
M. Gao, J. Skolnick, A threading-based method for the prediction of DNA-binding proteins with application to the human genome, PLoS Comput. Biol., 5 (2009), e1000567. https://doi.org/10.1371/journal.pcbi.1000567 doi: 10.1371/journal.pcbi.1000567
|
| [31] | X. Du, Y. Diao, H. Liu, S. Li, MsDBP: Exploring DNA-binding Proteins by Integrating Multi-scale Sequence Information via Chou's 5-steps Rule, J. Proteome Res., 18 (2019). https://doi.org/10.1021/acs.jproteome.9b00226 |
| [32] |
W. Lu, X. Chen, Y. Zhang, H. Wu, Y. Ding, J. Shen, et al., Research on DNA-binding protein identification method based on LSTM-CNN feature fusion, Comput. Math. Methods Med., 2022 (2022). https://doi.org/10.1155/2022/9705275 doi: 10.1155/2022/9705275
|
| [33] |
W. Lu, N. Zhou, Y. Ding, H. Wu, Y. Zhang, Q. Fu, et al., Application of DNA-binding protein prediction based on graph convolutional network and contact map, Biomed Res. Int., 2022 (2022). https://doi.org/10.1155/2022/9044793 doi: 10.1155/2022/9044793
|
| [34] |
M. Michel, D. Menéndez Hurtado, A. Elofsson, PconsC4: fast, accurate and hassle-free contact predictions, Bioinformatics, 35 (2019), 2677–2679. https://doi.org/10.1093/bioinformatics/bty1036 doi: 10.1093/bioinformatics/bty1036
|
| [35] |
J. Yan, T. Jiang, J. Liu, Y. Lu, S. Guan, H. Li, et al., DNA-binding protein prediction based on deep transfer learning, Math. Biosci. Eng., 19 (2022), 7719–7736. https://doi.org/10.3934/mbe.2022362 doi: 10.3934/mbe.2022362
|
| [36] |
G. Li, X. Du, X. Li, L. Zou, G. Zhang, Z. Wu, Prediction of DNA binding proteins using local features and long-term dependencies with primary sequences based on deep learning, PeerJ, 9 (2021), e11262. https://doi.org/10.7717/peerj.11262 doi: 10.7717/peerj.11262
|
| [37] |
O. Barukab, F. Ali, W. Alghamdi, Y. Bassam, S. A. Khan, DBP-CNN: Deep learning-based prediction of DNA-binding proteins by coupling discrete cosine transform with two-dimensional convolutional neural network, Expert Syst. Appl., 197 (2022), 116729. https://doi.org/10.1016/j.eswa.2022.116729 doi: 10.1016/j.eswa.2022.116729
|
| [38] |
S. Guan, Y. Qian, T. Jiang, Y. Ding, M. Jiang, H. Wu, MV-H-RKM: A Multiple View-based Hypergraph Regularized Restricted Kernel Machine for predicting DNA-binding proteins, IEEE/ACM Trans. Comput. Biol. Bioinf., 20 (2022), 1246–1256. https://doi.org/10.1109/TCBB.2022.3183191 doi: 10.1109/TCBB.2022.3183191
|
| [39] |
Y. Ding, J. Tang, F. Guo, Identification of drug-target interactions via Dual Laplacian Regularized Least Squares with Multiple Kernel Fusion, Knowledge-Based Syst., 204 (2020), 106254. https://doi.org/10.1016/j.knosys.2020.106254 doi: 10.1016/j.knosys.2020.106254
|
| [40] |
Y. Ding, J. Tang, F. Guo, Identification of drug-side effect association via semisupervised model and multiple kernel learning, IEEE J. Biomed. Health Inf., 23 (2018), 2619–2632. https://doi.org/10.1109/JBHI.2018.2883834 doi: 10.1109/JBHI.2018.2883834
|
| [41] |
H. Yang, Y. Ding, J. Tang, F. Guo, Drug-disease associations prediction via multiple kernel-based dual graph regularized least squares, Appl. Soft Comput., 112 (2021), 107811. https://doi.org/10.1016/j.asoc.2021.107811 doi: 10.1016/j.asoc.2021.107811
|
| [42] |
X. Guo, P. Tiwari, Q. Zou, Y. Ding, Subspace projection-based weighted echo state networks for predicting therapeutic peptides, Knowledge-Based Syst., 263 (2023), 110307. https://doi.org/10.1016/j.knosys.2023.110307 doi: 10.1016/j.knosys.2023.110307
|
| [43] |
C. Cao, J. Wang, D. Kwok, F. Cui, Z. Zhang, D. Zhao, et al., webTWAS: a resource for disease candidate susceptibility genes identified by transcriptome-wide association study, Nucleic Acids Res., 50 (2022), D1123–D1130. https://doi.org/10.1093/nar/gkab957 doi: 10.1093/nar/gkab957
|
| [44] |
C. Cao, B. Ding, Q. Li, D. Kwok, J. Wu, Q. Long, Power analysis of transcriptome-wide association study: Implications for practical protocol choice, PLos Genet., 17 (2021), e1009405. https://doi.org/10.1371/journal.pgen.1009405 doi: 10.1371/journal.pgen.1009405
|
| [45] |
Z. Zhang, F. Cui, C. Cao, Q. Wang, Q. Zou, Single-cell RNA analysis reveals the potential risk of organ-specific cell types vulnerable to SARS-CoV-2 infections, Comput. Biol. Med., 140 (2022), 105092. https://doi.org/10.1016/j.compbiomed.2021.105092 doi: 10.1016/j.compbiomed.2021.105092
|
| [46] |
F. Cui, Z. Zhang, Q. Zou, Sequence representation approaches for sequence-based protein prediction tasks that use deep learning, Briefings Funct. Genomics, 20 (2021), 61–73. https://doi.org/10.1093/bfgp/elaa030 doi: 10.1093/bfgp/elaa030
|
| [47] | Y. Cai, S. L. Lin, Support vector machines for predicting rRNA-, RNA-, and DNA-binding proteins from amino acid sequence, Biochim. Biophys. Acta, Proteins Proteomics, 1648 (2003), 127–133. https://doi.org/10.1016/S1570-9639(03)00112-2 |
| [48] | Z. You, L. Zhu, C. Zheng, H. Yu, S. Deng, Z. Ji, Prediction of protein-protein interactions from amino acid sequences using a novel multi-scale continuous and discontinuous feature set, BMC Bioinf., 15 (2014). https://doi.org/10.1186/1471-2105-15-S15-S9 |
| [49] |
Z. P. Feng, C. T. Zhang, Prediction of membrane protein types based on the hydrophobic index of amino acids, J. Protein Chem., 19 (2000), 269–275. https://doi.org/10.1023/A:1007091128394 doi: 10.1023/A:1007091128394
|
| [50] |
L. Nanni, S. Brahnam, A. Lumini, Wavelet images and Chou's pseudo amino acid composition for protein classification, Amino Acids, 43 (2012), 657–665. https://doi.org/10.1007/s00726-011-1114-9 doi: 10.1007/s00726-011-1114-9
|
| [51] |
J. Jeong, X. Lin, X. Chen, On position-specific scoring matrix for protein function prediction, IEEE/ACM Trans. Comput. Biol. Bioinf., 8 (2011), 308–315. https://doi.org/10.1109/TCBB.2010.93 doi: 10.1109/TCBB.2010.93
|
| [52] |
K. C. Chou, H. B. Shen, MemType-2L: A Web server for predicting membrane proteins and their types by incorporating evolution information through Pse-PSSM, Biochem. Biophys. Res. Commun., 360 (2007), 339–345. https://doi.org/10.1016/j.bbrc.2007.06.027 doi: 10.1016/j.bbrc.2007.06.027
|
| [53] |
R. Xu, J. Zhou, H. Wang, Y. He, X. Wang, B. Liu, Identifying DNA-binding proteins by combining support vector machine and PSSM distance transformation, BMC Syst. Biol., 9 (2015), S10. https://doi.org/10.1186/1752-0509-9-S1-S10 doi: 10.1186/1752-0509-9-S1-S10
|
| [54] | B. Liu, F. Liu, X. Wang, J. Chen, L. Fang, C. K. Chen, Pse-in-One: a web server for generating various modes of pseudo components of DNA, RNA, and protein sequences, Nucleic Acids Res., 43 (2015), W65–W71. https://doi.org/10.1093/nar/gkv458 |
| [55] |
L. Houthuys, J. Suykens, Tensor-based restricted kernel machines for multi-view classification, Inf. Fusion, 68 (2021), 54–66. https://doi.org/10.1016/j.inffus.2020.10.022 doi: 10.1016/j.inffus.2020.10.022
|
| [56] |
J. Suykens, Deep restricted kernel machines using conjugate feature duality, Neural Comput., 29 (2017), 2123–2163. https://doi.org/10.1162/neco_a_00984 doi: 10.1162/neco_a_00984
|
| [57] | Y. Ding, W. He, J. Tang, Q. Zou, F. Guo, Laplacian regularized sparse representation based classifier for identifying DNA N4-methylcytosine sites via L2, 1/2-matrix norm, IEEE/ACM Trans. Comput. Biol. Bioinf., 20 (2023), 500–511. https://doi.org/10.1109/TCBB.2021.3133309 |
| [58] |
C. Ai, P. Tiwari, H. Yang, Y. Ding, J. Tang, F. Guo, Identification of DNA N4-methylcytosine sites via multi-view kernel sparse representation model, IEEE Trans. Artif. Intell., 2022 (2022), 1–10. https://doi.org/10.1109/TAI.2022.3187060 doi: 10.1109/TAI.2022.3187060
|
| [59] |
Y. Qian, Y. Ding, Q. Zou, F. Guo, Multi-view kernel sparse representation for identification of membrane protein types, IEEE/ACM Trans. Comput. Biol. Bioinf., 20 (2022), 1234–1245. https://doi.org/10.1109/TCBB.2022.3191325 doi: 10.1109/TCBB.2022.3191325
|
| [60] |
Y. Ding, P. Tiwari, Q. Zou, F. Guo, H. M. Pandey, C-loss based higher order fuzzy inference systems for identifying DNA N4-methylcytosine sites, IEEE Trans. Fuzzy Syst., 30 (2022), 4754–4765. https://doi.org/10.1109/TFUZZ.2022.3159103 doi: 10.1109/TFUZZ.2022.3159103
|
| [61] |
Y. Ding, P. Tiwari, F. Guo, Q. Zou, Shared subspace-based radial basis function neural network for identifying ncRNAs subcellular localization, Neural Networks, 156 (2022), 170–178. https://doi.org/10.1016/j.neunet.2022.09.026 doi: 10.1016/j.neunet.2022.09.026
|
| [62] |
T. Wang, L. Zhang, W. Hu, Bridging deep and multiple kernel learning: A review, Inf. Fusion, 67 (2021), 3–13. https://doi.org/10.1016/j.inffus.2020.10.002 doi: 10.1016/j.inffus.2020.10.002
|
| [63] |
Y. Ding, J. Tang, F. Guo, Identification of drug-side effect association via semi-supervised model and multiple kernel learning, IEEE J. Biomed. Health Inf., 23 (2018), 2619–2632. https://doi.org/10.1109/JBHI.2018.2883834 doi: 10.1109/JBHI.2018.2883834
|
| [64] |
Y. Qian, Y. Ding, Q. Zou, F. Guo, Identification of drug-side effect association via restricted Boltzmann machines with penalized term, Briefings Bioinf., 23 (2022), bbac458. https://doi.org/10.1093/bib/bbac458 doi: 10.1093/bib/bbac458
|
| [65] |
Y. Ding, J. Tang, F. Guo, Identification of drug-side effect association via multiple information integration with centered kernel alignment, Neurocomputing, 325 (2019), 211–224. https://doi.org/10.1016/j.neucom.2018.10.028 doi: 10.1016/j.neucom.2018.10.028
|
| [66] |
Y. Wang, X. Liu, Y. Dou, Q. Lv, Y. Lu, Multiple kernel learning with hybrid kernel alignment maximization, Pattern Recognit., 70 (2017), 104–111. https://doi.org/10.1016/j.patcog.2017.05.005 doi: 10.1016/j.patcog.2017.05.005
|
| [67] |
J. O. Agushaka, A. E. Ezugwu, L. Abualigah, Dwarf mongoose optimization algorithm, Comput. Methods Appl. Mech. Eng., 391 (2022), 114570. https://doi.org/10.1016/j.cma.2022.114570 doi: 10.1016/j.cma.2022.114570
|
| [68] |
L. Abualigah, D. Yousri, M. A. Elaziz, A. A. Ewees, M. A. A. Al-Qaness, A. H. Gandomi, Aquila optimizer: a novel meta-heuristic optimization algorithm, Comput. Ind. Eng., 157 (2021), 107250. https://doi.org/10.1016/j.cie.2021.107250 doi: 10.1016/j.cie.2021.107250
|
| [69] |
L. Abualigah, M. A. Elaziz, P. Sumari, Z. W. Geem, A. H. Gandomi, Reptile Search Algorithm (RSA): A nature-inspired meta-heuristic optimizer, Expert Syst. Appl., 191 (2022), 116158. https://doi.org/10.1016/j.eswa.2021.116158 doi: 10.1016/j.eswa.2021.116158
|
| [70] |
O. N. Oyelade, A. E. Ezugwu, T. I. A. Mohamed, L. Abualigah, Ebola optimization search algorithm: A new nature-inspired metaheuristic optimization algorithm, IEEE Access, 10 (2022), 16150–16177. https://doi.org/10.1109/ACCESS.2022.3147821 doi: 10.1109/ACCESS.2022.3147821
|
| [71] |
L. Abualigah, A. Diabat, S. Mirjalili, M. A. Elaziz, A. H. Gandomi, The arithmetic optimization algorithm, Comput. Methods Appl. Mech. Eng., 376 (2021), 113609. https://doi.org/10.1016/j.cma.2020.113609 doi: 10.1016/j.cma.2020.113609
|
| [72] |
L. Abualigah, A. Diabat, P. Sumari, A. H. Gandomi, Applications, deployments, and integration of internet of drones (IoD): a review, IEEE Sens. J., 21 (2021), 25532–25546. https://doi.org/10.1109/JSEN.2021.3114266 doi: 10.1109/JSEN.2021.3114266
|
| [73] | M. Grant, S. Boyd, Y. Ye, CVX: Matlab Software For Disciplined Convex Programming, 2011. |
| [74] | L. Houthuys, Z. Karevan, J. Suykens, Multi-view LS-SVM regression for black-box temperature prediction in weather forecasting, in 2017 International Joint Conference on Neural Networks (IJCNN), 2017. https://doi.org/10.1109/IJCNN.2017.7965975 |
| [75] |
L. Cheng, Y. Hu, J. Sun, M. Zhou, Q. Jiang, DincRNA: a comprehensive web-based bioinformatics toolkit for exploring disease associations and ncRNA function, Bioinformatics, 34 (2018), 1953–1956. https://doi.org/10.1093/bioinformatics/bty002 doi: 10.1093/bioinformatics/bty002
|
| [76] |
N. Q. K. Le, Q. Ho, V. Nguyen, J. Chang, BERT-Promoter: An improved sequence-based predictor of DNA promoter using BERT pre-trained model and SHAP feature selection, Comput. Biol. Chem., 99 (2022), 107732. https://doi.org/10.1016/j.compbiolchem.2022.107732 doi: 10.1016/j.compbiolchem.2022.107732
|
| [77] |
N. Q. K. Le, D. T. Do, Q. A. Le, A sequence-based prediction of Kruppel-like factors proteins using XGBoost and optimized features, Gene, 787 (2021), 145643. https://doi.org/10.1016/j.gene.2021.145643 doi: 10.1016/j.gene.2021.145643
|
| [78] | T. Chen, C. Guestrin, Xgboost: A scalable tree boosting system, in Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, (2016), 785–794. https://doi.org/10.1145/2939672.2939785 |
| [79] |
L. Breiman, Random forests, Mach. Learn., 45 (2001), 5–32. https://doi.org/10.1023/A:1010933404324 doi: 10.1023/A:1010933404324
|
| [80] | G. Guo, H. Wang, D. Bell, Y. Bi, K. Greer, KNN model-based approach in classification, in On The Move to Meaningful Internet Systems 2003: CoopIS, DOA, and ODBASE. OTM 2003. Lecture Notes in Computer Science, Springer, Berlin, Heidelberg, 2888 (2003). https://doi.org/10.1007/978-3-540-39964-3_62 |
| [81] |
H. Wang, Y. Ding, J. Tang, F. Guo, Identification of membrane protein types via multivariate information fusion with Hilbert-Schmidt Independence Criterion, Neurocomputing, 383 (2020), 257–269. https://doi.org/10.1016/j.neucom.2019.11.103 doi: 10.1016/j.neucom.2019.11.103
|
| [82] | J. He, S. Chang, L. Xie, Fast kernel learning for spatial pyramid matching, in 2008 IEEE Conference on Computer Vision and Pattern Recognition, IEEE, 2008. https://doi.org/10.1109/CVPR.2008.4587636 |
| [83] |
W. Lin, J. Fang, X. Xiao, K. Chou, iDNA-Prot: Identification of DNA binding proteins using random forest with grey model, PLOS ONE, 6 (2011), e24756. https://doi.org/10.1371/journal.pone.0024756 doi: 10.1371/journal.pone.0024756
|
| [84] |
B. Liu, J. Xu, X. Lan, R. Xu, J. Zhou, X. Wang, et al., iDNA-Prot|dis: Identifying DNA-binding proteins by incorporating amino acid distance-pairs and reduced alphabet profile into the general pseudo amino acid composition, PLOS ONE, 9 (2014), e106691. https://doi.org/10.1371/journal.pone.0106691 doi: 10.1371/journal.pone.0106691
|
| [85] |
L. Wei, J. Tang, Z. Quan, Local-DPP: An improved DNA-binding protein prediction method by exploring local evolutionary information, Inf. Sci., 384 (2016), 135–144. https://doi.org/10.1016/j.ins.2016.06.026 doi: 10.1016/j.ins.2016.06.026
|
| [86] |
Y. Ding, F. Chen, X. Guo, J. Tang, H. Wu, Identification of DNA-binding proteins by multiple kernel support vector machine and sequence information, Curr. Proteomics, 17 (2020), 302–310. https://doi.org/10.2174/1570164616666190417100509 doi: 10.2174/1570164616666190417100509
|
| [87] |
Y. Zou, Y. Ding, J. Tang, F. Guo, L. Peng, FKRR-MVSF: A fuzzy kernel ridge regression model for identifying DNA-binding proteins by multi-view sequence features via Chou's five-step rule, Int. J. Mol. Sci., 20 (2019), 4175. https://doi.org/10.3390/ijms20174175 doi: 10.3390/ijms20174175
|
| [88] |
Y. Zou, H. Wu, X. Guo, L. Peng, Y. Ding, J. Tang, et al., MK-FSVM-SVDD: a multiple kernel-based fuzzy SVM model for predicting DNA-binding proteins via support vector data description, Curr. Bioinf., 16 (2021), 274–283. https://doi.org/10.2174/1574893615999200607173829 doi: 10.2174/1574893615999200607173829
|
| [89] |
J. Wang, S. Zhang, H. Qiao, J. Wang, UMAP-DBP: an improved DNA-binding proteins prediction method based on uniform manifold approximation and projection, Protein J., 40 (2021), 562–575. https://doi.org/10.1007/s10930-021-10011-y doi: 10.1007/s10930-021-10011-y
|






Figures(7) / Tables(10)

Yuqing Qian, Tingting Shang, Fei Guo, Chunliang Wang, Zhiming Cui, Yijie Ding, Hongjie Wu. Identification of DNA-binding protein based multiple kernel model[J]. Mathematical Biosciences and Engineering, 2023, 20(7): 13149-13170. doi: 10.3934/mbe.2023586







 DownLoad:
DownLoad: