Although multiple hub genes have been identified in head and neck squamous cell cancer (HNSCC) in recent years, because of the limited sample size and inconsistent bioinformatics analysis methods, the results are not reliable. Therefore, it is urgent to use reliable algorithms to find new prognostic markers of HNSCC.
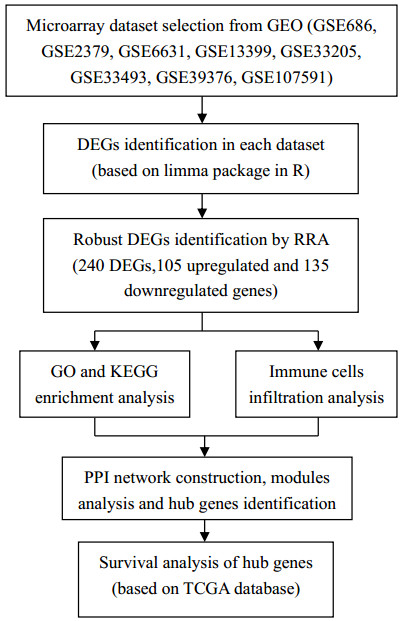
The Robust Rank Aggregation (RRA) method was used to integrate 8 microarray datasets of HNSCC downloaded from the Gene Expression Omnibus (GEO) database to screen differentially expressed genes (DEGs). Later, Gene Ontology (GO) functional annotation together with Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis was carried out to discover functions of those discovered DEGs. According to the KEGG results, those discovered DEGs showed tight association with the occurrence and development of HNSCC. Then cibersort algorithm was used to analyze the infiltration of immune cells of HNSCC and we found that the main infiltrated immune cells were B cells, dendritic cells and macrophages. A protein-protein interaction (PPI) network was established; moreover, key modules were also constructed to select 5 hub genes from the whole network using cytoHubba. 3 hub genes showed significant relationship with prognosis for TCGA-derived HNSCC patients.
The potent DEGs along with hub genes were selected by the combined bioinformatic approach. AURKA, BIRC5 and UBE2C genes may be the potential prognostic biomarker and therapeutic targets of HNSCC.
The Robust Rank Aggregation method and cibersort algorithm method can accurately predict the potential prognostic biomarker and therapeutic targets of HNSCC through multiple GEO datasets.
Citation: Tingting Chen, Wei Hua, Bing Xu, Hui Chen, Minhao Xie, Xinchen Sun, Xiaolin Ge. Robust rank aggregation and cibersort algorithm applied to the identification of key genes in head and neck squamous cell cancer[J]. Mathematical Biosciences and Engineering, 2021, 18(4): 4491-4507. doi: 10.3934/mbe.2021228
Although multiple hub genes have been identified in head and neck squamous cell cancer (HNSCC) in recent years, because of the limited sample size and inconsistent bioinformatics analysis methods, the results are not reliable. Therefore, it is urgent to use reliable algorithms to find new prognostic markers of HNSCC.
The Robust Rank Aggregation (RRA) method was used to integrate 8 microarray datasets of HNSCC downloaded from the Gene Expression Omnibus (GEO) database to screen differentially expressed genes (DEGs). Later, Gene Ontology (GO) functional annotation together with Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis was carried out to discover functions of those discovered DEGs. According to the KEGG results, those discovered DEGs showed tight association with the occurrence and development of HNSCC. Then cibersort algorithm was used to analyze the infiltration of immune cells of HNSCC and we found that the main infiltrated immune cells were B cells, dendritic cells and macrophages. A protein-protein interaction (PPI) network was established; moreover, key modules were also constructed to select 5 hub genes from the whole network using cytoHubba. 3 hub genes showed significant relationship with prognosis for TCGA-derived HNSCC patients.
The potent DEGs along with hub genes were selected by the combined bioinformatic approach. AURKA, BIRC5 and UBE2C genes may be the potential prognostic biomarker and therapeutic targets of HNSCC.
The Robust Rank Aggregation method and cibersort algorithm method can accurately predict the potential prognostic biomarker and therapeutic targets of HNSCC through multiple GEO datasets.

| [1] | R. L. Siegel, K. D. Miller, A. Jemal, Cancer statistics, CA Cancer J. Clin., 66 (2016), 7-30. |
| [2] | W. Chen, R. Zheng, P. D. Baade, S. Zhang, H. Zeng, F. Bray, et al., Cancer statistics in China, CA Cancer J. Clin., 66 (2016), 115-132. |
| [3] |
A. G. Sacco, E. E. Cohen, Current treatment options for recurrent or metastatic head and neck squamous cell carcinoma, J. Clin. Oncol., 33 (2015), 3305-3313. doi: 10.1200/JCO.2015.62.0963

|
| [4] | T. Y. Seiwert, B. Burtness, R. Mehra, J. Weiss, R. Berger, J. P. Eder, et al., Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial, Lancet Oncol., 17 (2016), 956-965. |
| [5] | R. L. Ferris, G. Blumenschein, J. Fayette, J. Guigay, A. D. Colevas, L. Licitra, et al., Nivolumab for recurrent squamous-cell carcinoma of the head and neck, N. Engl. J. Med., 375 (2016), 1856-1867. |
| [6] | Y. Li, X. Q. Tang, Z. Bai, X. Dai, Exploring the intrinsic differences among breast tumor subtypes defined using immunohistochemistry markers based on the decision tree, Sci. Rep., 6 (2016), 35773. |
| [7] |
R. Kolde, S. Laur, P. Adler, J. Vilo, Robust rank aggregation for gene list integration and meta-analysis, Bioinformatics, 28 (2012), 573-580. doi: 10.1093/bioinformatics/bts520
|
| [8] | Z. Y. Song, F. Chao, Z. Zhuo, Z. Ma, W. Li, G. Chen, Identification of hub genes in prostate cancer using robust rank aggregation and weighted gene co-expression network analysis, Aging (Albany N. Y.), 11 (2019), 4736-4756. |
| [9] | Y. Yang, Q. Lu, X. Shao, B. Mo, X. Nie, W. Liu, et al., Development of a three-gene prognostic signature for hepatitis B virus associated hepatocellular carcinoma based on integrated transcriptomic analysis, J. Cancer, 9 (2018), 1989-2002. |
| [10] |
D. Y. Zhuang, L. Jiang, Q. Q. He, P. Zhou, T. Yue, Identification of hub subnetwork based on topological features of genes in breast cancer, Int. J. Mol. Med., 35 (2015), 664-674. doi: 10.3892/ijmm.2014.2057
|
| [11] | B. Jin, W. Wang, G. Du, G. Z. Huang, L. T. Han, Z. Y. Tang, Identifying hub genes and dysregulated pathways in hepatocellular carcinoma, Eur. Rev. Med. Pharmacol. Sci., 19 (2015), 592-601. |
| [12] | W. Chang, L. Ma, L. Lin, L. Gu, X. Liu, H. Cai, et al., Identification of novel hub genes associated with liver metastasis of gastric cancer, Int. J. Cancer, 125 (2009), 2844-2853. |
| [13] | C. H. Chung, J. S. Parker, G. Karaca, J. Wu, W. K. Funkhouser, D. Moore, et al., Molecular classification of head and neck squamous cell carcinomas using patterns of gene expression, Cancer Cell, 5 (2004), 489-500. |
| [14] | A. Cromer, A. Carles, R. Millon, G. Ganguli, F. Chalmel, F. Lemaire, et al., Identification of genes associated with tumorigenesis and metastatic potential of hypopharyngeal cancer by microarray analysis, Oncogene, 23 (2004), 2484-2498. |
| [15] | M. A. Kuriakose, W. T. Chen, Z. M. He, A. G. Sikora, P. Zhang, Z. Y. Zhang, et al., Selection and validation of differentially expressed genes in head and neck cancer, Cell. Mol. Life Sci., 61 (2004), 1372-1383. |
| [16] | C. R. Cabanski, Y. Qi, X. Yin, E. Bair, M. C. Hayward, C. Fan, et al., Swiss made: Standardized within class sum of squares to evaluate methodologies and dataset elements, PLoS One, 5 (2010), e9905. |
| [17] | W. Sun, D. A. Gaykalova, M. F. Ochs, E. Mambo, D. Arnaoutakis, Y. Liu, et al., Activation of the NOTCH pathway in head and neck cancer, Cancer Res., 74 (2014), 1091-1104. |
| [18] | J. C. Stansfield, M. Rusay, R. Shan, C. Kelton, D. A. Gaykalova, E. J. Fertig, et al., Toward signaling-driven biomarkers immune to normal tissue contamination, Cancer Inform., 15 (2016), 15-21. |
| [19] | L. Verduci, M. Ferraiuolo, A. Sacconi, F. Ganci, J. Vitale, T. Colombo, et al., The oncogenic role of circPVT1 in head and neck squamous cell carcinoma is mediated through the mutant p53/YAP/TEAD transcription-competent complex, Genome Biol., 18 (2017), 237. |
| [20] | A. Sacconi, S. Donzelli, C. Pulito, S. Ferrero, F. Spinella, A. Morrone, et al., TMPRSS2, a SARS-CoV-2 internalization protease is downregulated in head and neck cancer patients, J. Exp. Clin. Cancer Res., 39 (2020), 200. |
| [21] | A. M. Newman, C. L. Liu, M. R. Green, A. J. Gentles, W. Feng, Y. Xu, et al., Robust enumeration of cell subsets from tissue expression profiles, Nat. Methods, 12 (2015), 453-457. |
| [22] | C. H. Chin, S. H. Chen, H. H. Wu, C. W. Ho, M. T. Ko, C. Y. Lin, cytoHubba: identifying hub objects and sub-networks from complex interactome, BMC Syst. Biol., 8 (2014), S11. |
| [23] | Y. Ding, M. Li, T. Tayier, L. Chen, S. Feng, Identification of novel prognostic biomarkers in head and neck squamous cell carcinoma using bioinformatics analysis, Comb. Chem. High Throughput Screen., 2020. |
| [24] | C. M. C. Petronacci, A. G. Garcia, E. P. Iruegas, B. R. Mundina, A. I. L. Pouso, M. P. Sayans, Identification of prognosis associated microRNAs in HNSCC subtypes based on TCGA dataset, Medicina (Kaunas), 56 (2020). |
| [25] |
J. P. Thiery, H. Acloque, R. Y. Huang, M. A. Nieto, Epithelial-mesenchymal transitions in development and disease, Cell, 139 (2009), 871-890. doi: 10.1016/j.cell.2009.11.007
|
| [26] |
K. Kessenbrock, V. Plaks, Z. Werb, Matrix metalloproteinases: regulators of the tumor microenvironment, Cell, 141 (2010), 52-67. doi: 10.1016/j.cell.2010.03.015
|
| [27] |
P. Lu, V. M. Weaver, Z. Werb, The extracellular matrix: a dynamic niche in cancer progression, J. Cell Biol., 196 (2012), 395-406. doi: 10.1083/jcb.201102147
|
| [28] | S. Furuta, Y. M. Jeng, L. Zhou, L. Huang, I. Kuhn, M. J. Bissell, et al., IL-25 causes apoptosis of IL-25R-expressing breast cancer cells without toxicity to nonmalignant cells, Sci. Transl. Med., 3 (2011), 7831. |
| [29] | C. K. Huang, C. Y. Yang, Y. M. Jeng, C. L. Chen, H. H. Wu, Y. C. Chang, et al., Autocrine/paracrine mechanism of interleukin-17B receptor promotes breast tumorigenesis through NF-kappaB-mediated antiapoptotic pathway, Oncogene, 33 (2014), 2968-2977. |
| [30] | Q. Bie, P. Zhang, Z. Su, D. Zheng, X. Ying, Y. Wu, et al., Polarization of ILC2s in peripheral blood might contribute to immunosuppressive microenvironment in patients with gastric cancer, J. Immunol. Res., 2014 (2014), 923135. |
| [31] | A. Al-Samadi, S. Moossavi, A. Salem, M. Sotoudeh, S. M. Tuovinen, Y. T. Konttinen, et al., Distinctive expression pattern of interleukin-17 cytokine family members in colorectal cancer, Tumour. Biol., 37 (2016), 1609-1615. |
| [32] | Y. F. Yang, Y. C. Lee, S. Lo, Y. N. Chung, Y. C. Hsieh, W. C. Chiu, et al., A positive feedback loop of IL-17B-IL-17RB activates ERK/beta-catenin to promote lung cancer metastasis, Cancer Lett., 422 (2018), 44-55. |
| [33] |
B. B. Zhou, S. J. Elledge, The DNA damage response: putting checkpoints in perspective, Nature, 408 (2000), 433-439. doi: 10.1038/35044005
|
| [34] |
J. H. Hoeijmakers, Genome maintenance mechanisms for preventing cancer, Nature, 411 (2001), 366-374. doi: 10.1038/35077232
|
| [35] |
Y. Naito, Y. Yoshioka, Y. Yamamoto, T. Ochiya, How cancer cells dictate their microenvironment: present roles of extracellular vesicles, Cell. Mol. Life Sci., 74 (2017), 697-713. doi: 10.1007/s00018-016-2346-3
|
| [36] |
T. L. Whiteside, Exosome and mesenchymal stem cell cross-talk in the tumor microenvironment, Semin. Immunol., 35 (2018), 69-79. doi: 10.1016/j.smim.2017.12.003
|
| [37] |
M. De Palma, D. Biziato, T. V. Petrova, Microenvironmental regulation of tumour angiogenesis, Nat. Rev. Cancer, 17 (2017), 457-474. doi: 10.1038/nrc.2017.51
|
| [38] |
J. D. Hansen, L. D. Pasquier, M. P. Lefranc, V. Lopez, A. Benmansour, P. Boudinot, The B7 family of immunoregulatory receptors: a comparative and evolutionary perspective, Mol. Immunol., 46 (2009), 457-472. doi: 10.1016/j.molimm.2008.10.007
|
| [39] | M. Janakiram, U. A. Shah, W. Liu, A. Zhao, M. P. Schoenberg, X. Zang, The third group of the B7-CD28 immune checkpoint family: HHLA2, TMIGD2, B7x, and B7-H3, Immunol. Rev., 276 (2017), 26-39. |
| [40] |
L. Chen, T. Azuma, W. Yu, X. Zheng, L. Luo, L. Chen, B7-H1 maintains the polyclonal T cell response by protecting dendritic cells from cytotoxic T lymphocyte destruction, Proc. Natl. Acad. Sci. U. S. A., 115 (2018), 3126-3131. doi: 10.1073/pnas.1722043115
|
| [41] |
S. Dutertre, S. Descamps, C. Prigent, On the role of aurora-A in centrosome function, Oncogene, 21 (2002), 6175-6183. doi: 10.1038/sj.onc.1205775
|
| [42] | T. Marumoto, D. Zhang, H. Saya, Aurora-A-a guardian of poles, Nat. Rev. Cancer, 5 (2005), 42-50. |
| [43] | P. Cammareri, A. Scopelliti, M. Todaro, V. Eterno, F. Francescangeli, M. P. Moyer, et al., Aurora-a is essential for the tumorigenic capacity and chemoresistance of colorectal cancer stem cells, Cancer Res., 70 (2010), 4655-4665. |
| [44] | T. Tanaka, M. Kimura, K. Matsunaga, D. Fukada, H. Mori, Y. Okano, Centrosomal kinase AIK1 is overexpressed in invasive ductal carcinoma of the breast, Cancer Res., 59 (1999), 2041-2044. |
| [45] | D. Li, J. Zhu, P. F. Firozi, J. L. Abbruzzese, D. B. Evans, K. Cleary, et al., Overexpression of oncogenic STK15/BTAK/Aurora a kinase in human pancreatic cancer, Clin. Cancer Res., 9 (2003), 991-997. |
| [46] | K. Kamada, Y. Yamada, T. Hirao, H. Fujimoto, Y. Takahama, M. Ueno, et al., Amplification/overexpression of Aurora-A in human gastric carcinoma: potential role in differentiated type gastric carcinogenesis, Oncol. Rep., 12 (2004), 593-599. |
| [47] | S. B. Yang, X. B. Zhou, H. X. Zhu, L. P. Quan, J. F. Bai, J. He, et al., Amplification and overexpression of Aurora-A in esophageal squamous cell carcinoma, Oncol. Rep., 17 (2007), 1083-1088. |
| [48] | Y. Yang, L. Ding, Q. Zhou, L. Fen, Y. Cao, J. Sun, et al., Silencing of AURKA augments the antitumor efficacy of the AURKA inhibitor MLN8237 on neuroblastoma cells, Cancer Cell Int., 20 (2020), 9. |
| [49] |
A. Mesic, M. Rogar, P. Hudler, R. Juvan, R. Komel, Association of the AURKA and AURKC gene polymorphisms with an increased risk of gastric cancer, IUBMB Life, 68 (2016), 634-644. doi: 10.1002/iub.1521
|
| [50] | X. Liu, Z. Li, Y. Song, R. Wang, L. Han, Q. Wang, et al., AURKA induces EMT by regulating histone modification through Wnt/beta-catenin and PI3K/Akt signaling pathway in gastric cancer, Oncotarget, 7 (2016), 33152-33164. |
| [51] | M. Kamran, Z. J. Long, D. Xu, S. S. Lv, B. Liu, C. L. Wang, et al., Aurora kinase a regulates survivin stability through targeting FBXL7 in gastric cancer drug resistance and prognosis, Oncogenesis, 6 (2017), e298. |
| [52] |
A. A. Dar, A. Belkhiri, J. Ecsedy, A. Zaika, W. El-Rifai, Aurora kinase a inhibition leads to p73-dependent apoptosis in p53-deficient cancer cells, Cancer Res., 68 (2008), 8998-9004. doi: 10.1158/0008-5472.CAN-08-2658
|
| [53] | H. Katayama, J. Wang, W. Treekitkarnmongkol, H. Kawai, K. Sasai, H. Zhang, et al., Aurora kinase-A inactivates DNA damage-induced apoptosis and spindle assembly checkpoint response functions of p73, Cancer Cell, 21 (2012), 196-211. |
| [54] | V. Sehdev, A. Katsha, J. Arras, D. Peng, M. Soutto, J. Ecsedy, et al., HDM2 regulation by AURKA promotes cell survival in gastric cancer, Clin. Cancer Res., 20 (2014), 76-86. |
| [55] |
A. A. Dar, A. Belkhiri, W. El-Rifai, The aurora kinase a regulates GSK-3beta in gastric cancer cells, Oncogene, 28 (2009), 866-875. doi: 10.1038/onc.2008.434
|
| [56] | A. Katsha, M. Soutto, V. Sehdev, D. Peng, M. K. Washington, M. B. Piazuelo, et al., Aurora kinase a promotes inflammation and tumorigenesis in mice and human gastric neoplasia, Gastroenterology, 145 (2013), 1312-1322. |
| [57] |
B. S. Kochupurakkal, J. D. Iglehart, Identification of genes responsible for RelA-dependent proliferation arrest in human mammary epithelial cells conditionally expressing RelA, Genom. Data, 7 (2016), 92-93. doi: 10.1016/j.gdata.2015.11.022
|
| [58] | P. Gil-Kulik, A. Krzyzanowski, E. Dudzinska, J. Karwat, P. Chomik, M. Swistowska, et al., Potential involvement of BIRC5 in maintaining pluripotency and cell differentiation of human stem cells, Oxid. Med. Cell. Longev., 2019 (2019), 8727925. |
| [59] | C. Wang, X. Zheng, C. Shen, Y. Shi, MicroRNA-203 suppresses cell proliferation and migration by targeting BIRC5 and LASP1 in human triple-negative breast cancer cells, J. Exp. Clin. Cancer Res., 31 (2012), 58. |
| [60] | Q. Fu, J. Zhang, X. Xu, F. Qian, K. Feng, J. Ma, miR-203 is a predictive biomarker for colorectal cancer and its expression is associated with BIRC5, Tumour. Biol., 2016. |
| [61] | L. Cao, C. Li, S. Shen, Y. Yan, W. Ji, J. Wang, et al., OCT4 increases BIRC5 and CCND1 expression and promotes cancer progression in hepatocellular carcinoma, BMC Cancer, 13 (2013), 82. |
| [62] | I. Tamm, Y. Wang, E. Sausville, D. A. Scudiero, N. Vigna, T. Oltersdorf, et al., IAP-family protein survivin inhibits caspase activity and apoptosis induced by fas (CD95), bax, caspases, and anticancer drugs, Cancer Res., 58 (1998), 5315-5320. |
| [63] |
Y. David, T. Ziv, A. Admon, A. Navon, The E2 ubiquitin-conjugating enzymes direct polyubiquitination to preferred lysines, J. Biol. Chem., 285 (2010), 8595-8604. doi: 10.1074/jbc.M109.089003
|
| [64] |
Z. Ping, R. Lim, T. Bashir, M. Pagano, D. Guardavaccaro, APC/C (Cdh1) controls the proteasome-mediated degradation of E2F3 during cell cycle exit, Cell Cycle, 11 (2012), 1999-2005. doi: 10.4161/cc.20402
|
| [65] | K. Ieta, E. Ojima, F. Tanaka, Y. Nakamura, N. Haraguchi, K. Mimori, et al., Identification of overexpressed genes in hepatocellular carcinoma, with special reference to ubiquitin-conjugating enzyme E2C gene expression, Int. J. Cancer, 121 (2007), 33-38. |
| [66] | Y. Takahashi, Y. Ishii, Y. Nishida, M. Ikarashi, T. Nagata, T. Nakamura, et al., Detection of aberrations of ubiquitin-conjugating enzyme E2C gene (UBE2C) in advanced colon cancer with liver metastases by DNA microarray and two-color FISH, Cancer Genet. Cytogenet., 168 (2006), 30-35. |
| [67] | S. Z. Li, Y. Song, H. H. Zhang, B. X. Jin, Y. Liu, W. B. Liu, et al., UbcH10 overexpression increases carcinogenesis and blocks ALLN susceptibility in colorectal cancer, Sci. Rep., 4 (2014), 6910. |
| [68] | Z. Zhang, P. Liu, J. Wang, T. Gong, F. Zhang, J. Ma, et al., Ubiquitin-conjugating enzyme E2C regulates apoptosis-dependent tumor progression of non-small cell lung cancer via ERK pathway, Med. Oncol., 32 (2015), 149. |
| [69] |
Q. Han, C. Zhou, F. Liu, G. Xu, R. Zheng, X. Zhang, MicroRNA-196a post-transcriptionally upregulates the UBE2C proto-oncogene and promotes cell proliferation in breast cancer, Oncol. Rep., 34 (2015), 877-883. doi: 10.3892/or.2015.4049
|





Figures(6) / Tables(1)

Tingting Chen, Wei Hua, Bing Xu, Hui Chen, Minhao Xie, Xinchen Sun, Xiaolin Ge. Robust rank aggregation and cibersort algorithm applied to the identification of key genes in head and neck squamous cell cancer[J]. Mathematical Biosciences and Engineering, 2021, 18(4): 4491-4507. doi: 10.3934/mbe.2021228







 DownLoad:
DownLoad: