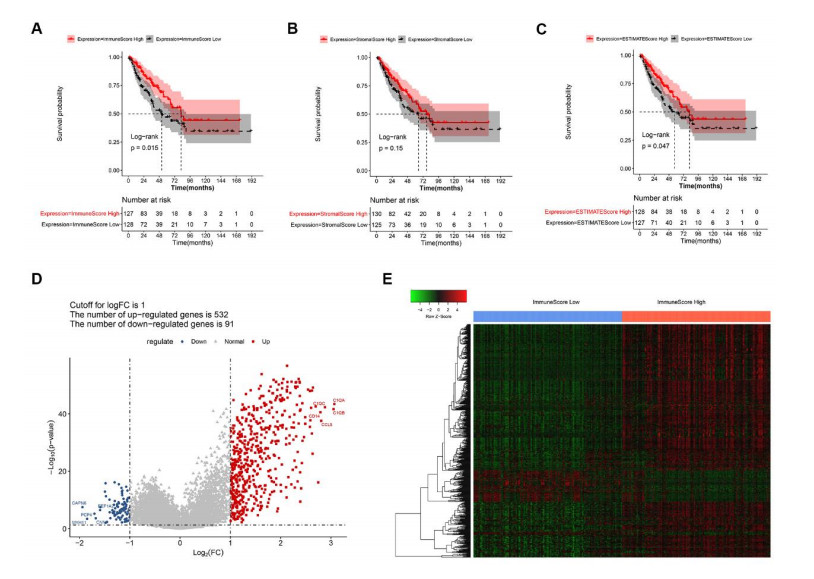
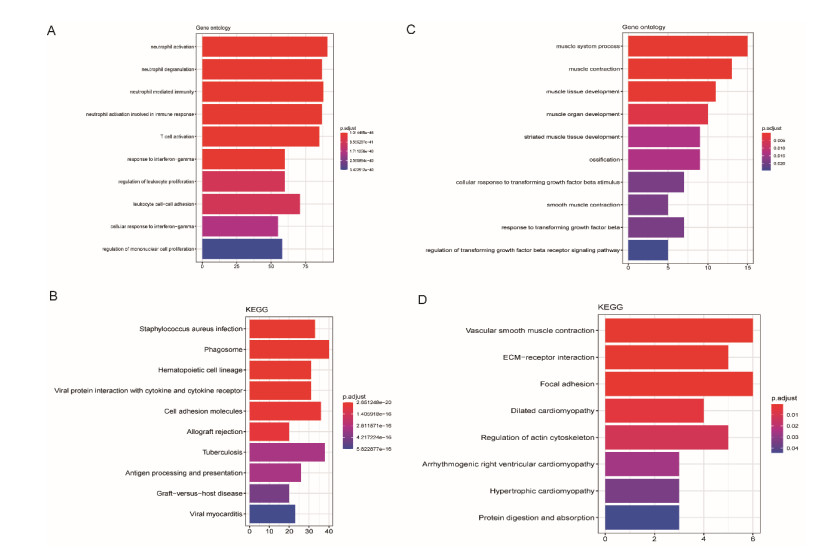
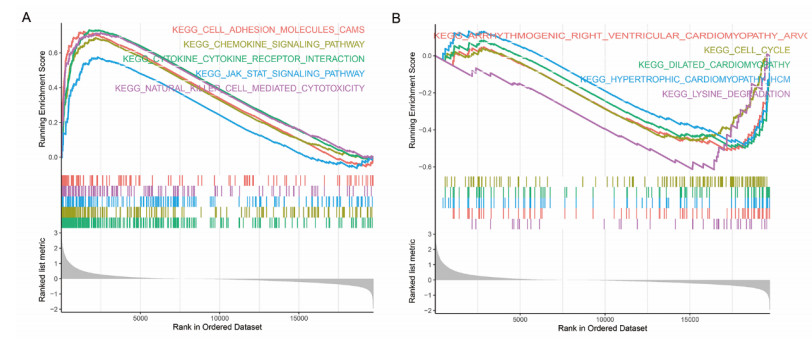
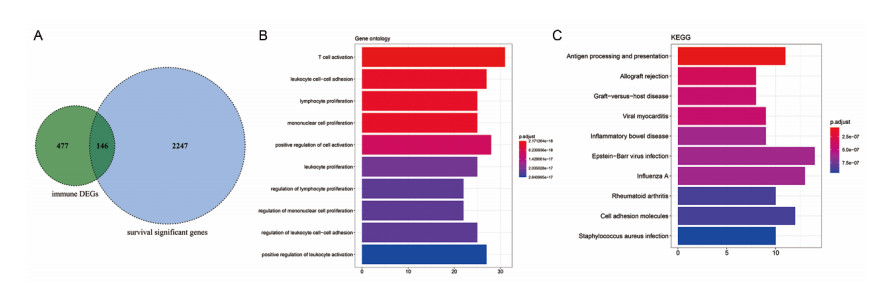
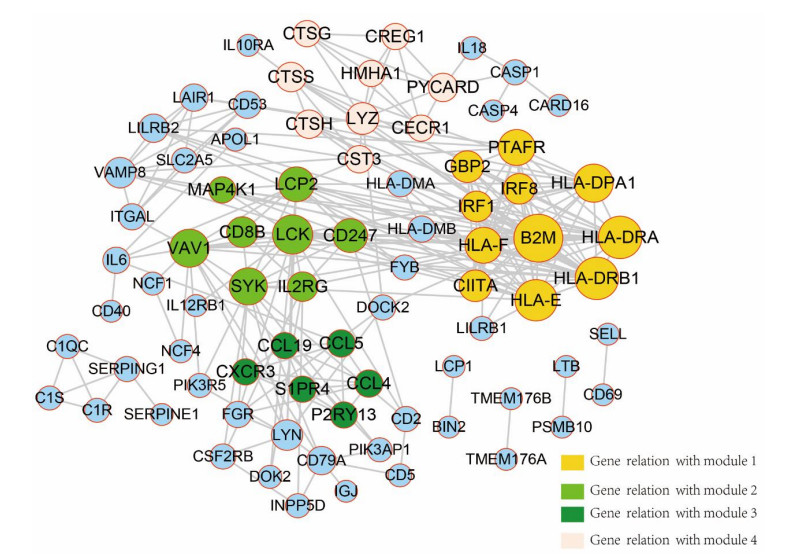
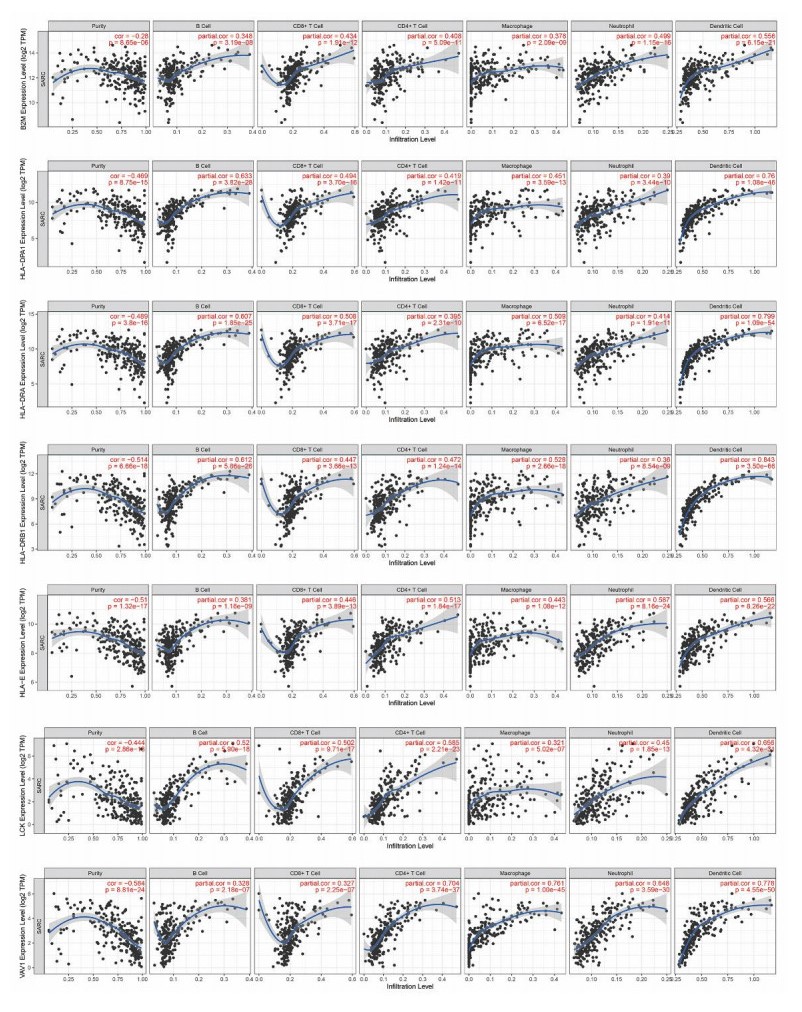
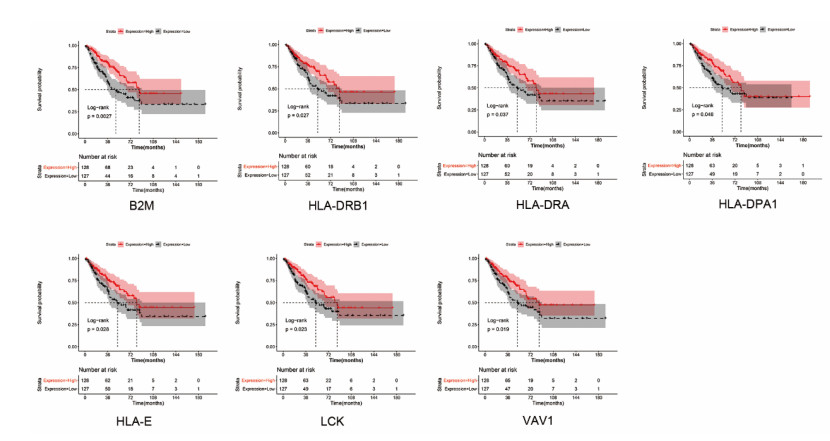
Sarcomas are a heterogeneous group of malignant mesenchymal neoplasms. This study aimed to investigate the immune-related prognostic gene signatures in the tumor microenvironment of sarcoma. The RNA-sequencing data and clinical phenotype data of 260 sarcoma samples and two normal samples were downloaded from The Cancer Genome Atla (TCGA) database. Tumor purity and immune cells infiltration were evaluated by Estimation of Stromal and Immune cells in Malignant Tumors using Expression data (ESTIMATE) deconvolution algorithm. Differentially expressed genes (DEGs) were screened in high vs. low immune score groups. Survival analysis was performed using Kaplan-Meier curve with log-rank test. Tumor infiltrating of immune cells was analyzed by Tumor Immune Estimation Resource (TIMER). High immune score and ESTIMATE score were associated with favorable prognosis. A total of 623 immune DEGs were screened. The majority of these genes (532 genes accounting for 85% of the DEGs) were up-regulated, and these genes were significantly enriched in various immune related biological processed and pathways, such as neutrophil activation, T cell activation, antigen processing and presentation. A total of 146 prognosis-related immune DEGs, and seven hub genes were identified, including B2M, HLA-DRB1, HLA-DRA, HLA-E, LCK, HLA-DPA1, and VAV1. Survival analysis showed that high expression of these genes was associated with a favorable prognosis. There were negative correlations between the expression of these hub genes and tumor purity, while positive correlations between expression of these hub genes and f infiltration levels of B cells, CD4+ T cells, CD8+ T cells, neutrophils, macrophages and dendritic cells. These results help to stratify patients with different immune subtypes and help to design immunotherapy strategies for these patients in sarcoma.
Citation: Jun Wang, Mingzhi Gong, Zhenggang Xiong, Yangyang Zhao, Deguo Xing. Immune-related prognostic genes signatures in the tumor microenvironment of sarcoma[J]. Mathematical Biosciences and Engineering, 2021, 18(3): 2243-2257. doi: 10.3934/mbe.2021113
Sarcomas are a heterogeneous group of malignant mesenchymal neoplasms. This study aimed to investigate the immune-related prognostic gene signatures in the tumor microenvironment of sarcoma. The RNA-sequencing data and clinical phenotype data of 260 sarcoma samples and two normal samples were downloaded from The Cancer Genome Atla (TCGA) database. Tumor purity and immune cells infiltration were evaluated by Estimation of Stromal and Immune cells in Malignant Tumors using Expression data (ESTIMATE) deconvolution algorithm. Differentially expressed genes (DEGs) were screened in high vs. low immune score groups. Survival analysis was performed using Kaplan-Meier curve with log-rank test. Tumor infiltrating of immune cells was analyzed by Tumor Immune Estimation Resource (TIMER). High immune score and ESTIMATE score were associated with favorable prognosis. A total of 623 immune DEGs were screened. The majority of these genes (532 genes accounting for 85% of the DEGs) were up-regulated, and these genes were significantly enriched in various immune related biological processed and pathways, such as neutrophil activation, T cell activation, antigen processing and presentation. A total of 146 prognosis-related immune DEGs, and seven hub genes were identified, including B2M, HLA-DRB1, HLA-DRA, HLA-E, LCK, HLA-DPA1, and VAV1. Survival analysis showed that high expression of these genes was associated with a favorable prognosis. There were negative correlations between the expression of these hub genes and tumor purity, while positive correlations between expression of these hub genes and f infiltration levels of B cells, CD4+ T cells, CD8+ T cells, neutrophils, macrophages and dendritic cells. These results help to stratify patients with different immune subtypes and help to design immunotherapy strategies for these patients in sarcoma.

| [1] |
C. A. Cipriano, E. Jang, W. Tyler, Sarcoma surveillance: A review of current evidence and guidelines, J. Am. Acad. Orthop. Surg., 28 (2020), 145-156. doi: 10.5435/JAAOS-D-19-00002

|
| [2] |
J. Y. Hui, Epidemiology and etiology of sarcomas, Surg. Clin. N. Am., 96 (2016), 901-914. doi: 10.1016/j.suc.2016.05.005
|
| [3] |
D. L. Kerr, B. L. Dial, A. L. Lazarides, A. A. Catanzano, W. O. Lane, D. G. Blazer, et al., Epidemiologic and survival trends in adult primary bone tumors of the spine, Spine J., 19 (2019), 1941-1949. doi: 10.1016/j.spinee.2019.07.003
|
| [4] |
F. Tang, E. Choy, C. Tu, F. Hornicek, Z. Duan, Therapeutic applications of histone deacetylase inhibitors in sarcoma, Cancer Treat. Rev., 59 (2017), 33-45. doi: 10.1016/j.ctrv.2017.06.006
|
| [5] |
B. T. Turner, L. Hampton, D. Schiller, L. A. Mack, C. Robertson-More, H. Li, et al., Neoadjuvant radiotherapy followed by surgery compared with surgery alone in the treatment of retroperitoneal sarcoma: A population-based comparison, Curr. Oncol., 26 (2019), e766-e772. doi: 10.3747/co.26.5185
|
| [6] |
A. Marabelle, L. Tselikas, T. de Baere, d R. Houot, Intratumoral immunotherapy: Using the tumor as the remedy, Ann. Oncol., 28 (2017), xii33-xii43. doi: 10.1093/annonc/mdx683
|
| [7] |
R. Shafabakhsh, M. H. Pourhanifeh, H. R. Mirzaei, A. Sahebkar, Z. Asemi, H. Mirzaei, Targeting regulatory T cells by curcumin: A potential for cancer immunotherapy, Pharmacol Res., 147 (2019), 104353. doi: 10.1016/j.phrs.2019.104353
|
| [8] |
M. Burgess, H. Tawbi, Immunotherapeutic approaches to sarcoma, Curr. Treat. Options Oncol., 16 (2015), 26. doi: 10.1007/s11864-015-0345-5
|
| [9] |
M. H. Gholamin, S. M. Razavi, S. M. Hassanian, L. Saadatpour, A. Masoudifar, S. ShahidSales, et al., GD2-targeted immunotherapy and potential value of circulating microRNAs in neuroblastoma, J. Cell. Physiol., 233 (2018), 866-879. doi: 10.1002/jcp.25793
|
| [10] |
F. Petitprez, A. de Reyniès, E. Z. Keung, T. W. Chen, C. M. Sun, J. Calderaro, et al., B cells are associated with survival and immunotherapy response in sarcoma, Nature, 577 (2020), 556-560. doi: 10.1038/s41586-019-1906-8
|
| [11] |
N. Oike, H. Kawashima, A. Ogose, T. Hotta, H. Hatano, T. Ariizumi, et al., Prognostic impact of the tumor immune microenvironment in synovial sarcoma, Cancer Sci., 109 (2018), 3043-3054. doi: 10.1111/cas.13769
|
| [12] |
M. Toulmonde, N. Penel, J. Adam, C. Chevreau, J. Y. Blay, A. Le Cesne, et al., Use of PD-1 targeting, macrophage infiltration, and IDO pathway activation in sarcomas: A phase 2 clinical trial, JAMA Oncol., 4 (2018), 93-97. doi: 10.1001/jamaoncol.2017.1617
|
| [13] |
P. Koirala, M. E. Roth, J. Gill, S. Piperdi, J. M. Chinai, D. S. Geller, et al., Immune infiltration and PD-L1 expression in the tumor microenvironment are prognostic in osteosarcoma, Sci. Rep., 6 (2016), 30093. doi: 10.1038/srep30093
|
| [14] |
C. Hu, B. Chen, Z. Huang, C. Liu, L. Ye, C. Wang, et al., Comprehensive profiling of immune-related genes in soft tissue sarcoma patients, J. Transl. Med., 18 (2020), 337. doi: 10.1186/s12967-020-02512-8
|
| [15] |
H. Y. Gu, L. L. Lin, C. Zhang, M. Yang, H. C. Zhong, R. X. Wei, The potential of five immune-related prognostic genes to predict survival and response to immune checkpoint inhibitors for soft tissue sarcomas based on multi-omic study, Front. Oncol., 10 (2020), 1317. doi: 10.3389/fonc.2020.01317
|
| [16] | L. Jafarzadeh, M. Khakpoor-Koosheh, H. Mirzaei, H. R. Mirzaei, Biomarkers for predicting the outcome of various cancer immunotherapies, Crit. Rev. Oncol. Hematol., 157 (2020), 103161. |
| [17] |
J. Harrow, A. Frankish, J. M. Gonzalez, E. Tapanari, M. Diekhans, F. Kokocinski, et al., GENCODE: The reference human genome annotation for The ENCODE Project, Genome Res., 22 (2012), 1760-1774. doi: 10.1101/gr.135350.111
|
| [18] |
K. Yoshihara, M. Shahmoradgoli, E. Martínez, R. Vegesna, H. Kim, W. Torres-Garcia, et al., Inferring tumour purity and stromal and immune cell admixture from expression data, Nat. Commun., 4 (2013), 2612. doi: 10.1038/ncomms3612
|
| [19] | G. K. Smyth limma: Linear Models for Microarray Data; In: R. CVJ Gentleman, W. Huber, R.A. Irizarry, S. Dudoit (eds), Springer, New York, NY. 2005. |
| [20] |
G. Yu, L. G. Wang, Y. Han, Q. Y. He, clusterProfiler: an R package for comparing biological themes among gene clusters, Omics, 16 (2012), 284-287. doi: 10.1089/omi.2011.0118
|
| [21] |
A. Liberzon, C. Birger, H. Thorvaldsdóttir, M. Ghandi, J. P. Mesirov, P. Tamayo, The Molecular Signatures Database (MSigDB) hallmark gene set collection, Cell Syst., 1 (2015), 417-425. doi: 10.1016/j.cels.2015.12.004
|
| [22] |
D. Szklarczyk, A. Franceschini, S. Wyder, K. Forslund, D. Heller, J. Huerta-Cepas, et al., STRING v10: Protein-protein interaction networks, integrated over the tree of life, Nucleic Acids Res., 43 (2015), D447-452. doi: 10.1093/nar/gku1003
|
| [23] |
P. Shannon, A. Markiel, O. Ozier, N. S. Baliga, J. T. Wang, D. Ramage, et al., Cytoscape: A software environment for integrated models of biomolecular interaction networks, Genome Res., 13 (2003), 2498-2504. doi: 10.1101/gr.1239303
|
| [24] |
W. P. Bandettini, P. Kellman, C. Mancini, O. J. Booker, S. Vasu, S. W. Leung, et al., MultiContrast Delayed Enhancement (MCODE) improves detection of subendocardial myocardial infarction by late gadolinium enhancement cardiovascular magnetic resonance: A clinical validation study, J. Cardiovasc. Magn. Reson., 14 (2012), 83. doi: 10.1186/1532-429X-14-83
|
| [25] |
T. Li, J. Fan, B. Wang, N. Traugh, Q. Chen, J. S. Liu, et al., TIMER: A web server for comprehensive analysis of tumor-infiltrating immune cells, Cancer Res., 77 (2017), e108-e110. doi: 10.1158/0008-5472.CAN-17-0307
|
| [26] |
Y. Yang, Cancer immunotherapy: Harnessing the immune system to battle cancer, J. Clin. Invest., 125 (2015), 3335-3337. doi: 10.1172/JCI83871
|
| [27] |
K. Tahkola, J. P. Mecklin, E. V. Wirta, M. Ahtiainen, O. Helminen, J. Böhm, et al., High immune cell score predicts improved survival in pancreatic cancer, Virchows Arch., 472 (2018), 653-665. doi: 10.1007/s00428-018-2297-1
|
| [28] |
B. Amulic, C. Cazalet, G. L. Hayes, K. D. Metzler, A. Zychlinsky, Neutrophil function: From mechanisms to disease, Annu. Rev. Immunol., 30 (2012), 459-489. doi: 10.1146/annurev-immunol-020711-074942
|
| [29] |
W. M. T. Kuwabara, J. Andrade-Silva, J. N. B. Pereira, J. H. Scialfa, J. Cipolla-Neto, Neutrophil activation causes tumor regression in Walker 256 tumor-bearing rats, Sci. Rep., 9 (2019), 16524. doi: 10.1038/s41598-019-52956-2
|
| [30] | S. Wang, Z. He, X. Wang, H. Li, X. S. Liu, Antigen presentation and tumor immunogenicity in cancer immunotherapy response prediction, Elife, 8 (2019). |
| [31] |
D. Chowell, L. G. T. Morris, C. M. Grigg, J. K. Weber, R. M. Samstein, V. Makarov, et al., Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy, Science, 359 (2018), 582-587. doi: 10.1126/science.aao4572
|
| [32] |
X. Bian, Y. T. Xiao, T. Wu, M. Yao, L. Du, S. Ren, et al., Microvesicles and chemokines in tumor microenvironment: mediators of intercellular communications in tumor progression, Mol. Cancer, 18 (2019), 50. doi: 10.1186/s12943-019-0973-7
|
| [33] |
K. Sun, C. Gong, H. Peng, H. Fang, J. Zhou, J. Li, et al., High CCL5 expression is associated with osteosarcoma metastasis and poor prognosis of patients with osteosarcoma, Mol. Med. Rep., 16 (2017), 6953-6957. doi: 10.3892/mmr.2017.7458
|
| [34] | Y. Tang, Z. Gu, Y. Fu, J. Wang, CXCR3 from chemokine receptor family correlates with immune infiltration and predicts poor survival in osteosarcoma, Biosci. Rep., 39 (2019). |
| [35] | S. Shahrabi, E. H. Hadad, A. A. Asnafi, M. M. Behzad, A. Ehsanpour, N. Saki, Human leukocyte antigens in cancer metastasis: Prognostic approach and therapeutic susceptibility, Histol. Histopathol., 34 (2019), 111-124. |
| [36] |
D. Berghuis, A. S. de Hooge, S. J. Santos, D. Horst, E. J. Wiertz, M. C. van Eggermond, et al., Reduced human leukocyte antigen expression in advanced-stage Ewing sarcoma: implications for immune recognition, J. Pathol., 218 (2009), 222-231. doi: 10.1002/path.2537
|
| [37] |
F. A. Leite, R. C. Lira, P. F. Fedatto, S. R. Antonini, C. E. Martinelli, Jr., M. de Castro, et al., Low expression of HLA-DRA, HLA-DPA1, and HLA-DPB1 is associated with poor prognosis in pediatric adrenocortical tumors (ACT), Pediatr. Blood Cancer, 61 (2014), 1940-1948. doi: 10.1002/pbc.25118
|
| [38] |
S. J. Luk, D. M. van der Steen, R. S. Hagedoorn, E. S. Jordanova, M. W. Schilham, J. V. Bovée, et al., PRAME and HLA Class I expression patterns make synovial sarcoma a suitable target for PRAME specific T-cell receptor gene therapy, Oncoimmunology, 7 (2018), e1507600. doi: 10.1080/2162402X.2018.1507600
|
| [39] |
A. Castro, K. Ozturk, R. M. Pyke, S. Xian, M. Zanetti, H. Carter, Elevated neoantigen levels in tumors with somatic mutations in the HLA-A, HLA-B, HLA-C and B2M genes, BMC Med. Genom., 12 (2019), 107. doi: 10.1186/s12920-019-0544-1
|
 mbe-18-03-113 Supplementary files.docx mbe-18-03-113 Supplementary files.docx |
 |

Figures(8)

Jun Wang, Mingzhi Gong, Zhenggang Xiong, Yangyang Zhao, Deguo Xing. Immune-related prognostic genes signatures in the tumor microenvironment of sarcoma[J]. Mathematical Biosciences and Engineering, 2021, 18(3): 2243-2257. doi: 10.3934/mbe.2021113







 DownLoad:
DownLoad: