To improve the understanding of the molecular mechanism of vitiligo is necessary to predict and formulate new targeted gene therapy strategies.
GSE65127, GSE75819, GSE53146 and GSE90880 were collected, and obtained four groups of differentially expressed genes (DEGs) by limma R package. Through weighted gene co-expression network analysis (WGCNA), the co-expression of genes with large variance in GSE65127 and GSE75819 was identified. Enrichment analysis of intersection gene between module genes and DEGs with the same up-regulated or down-regulated in GSE65127 and GSE75819 was performed. In addition, ssGSEA was used to identify the immune infiltration of vitiligo in four datasets.
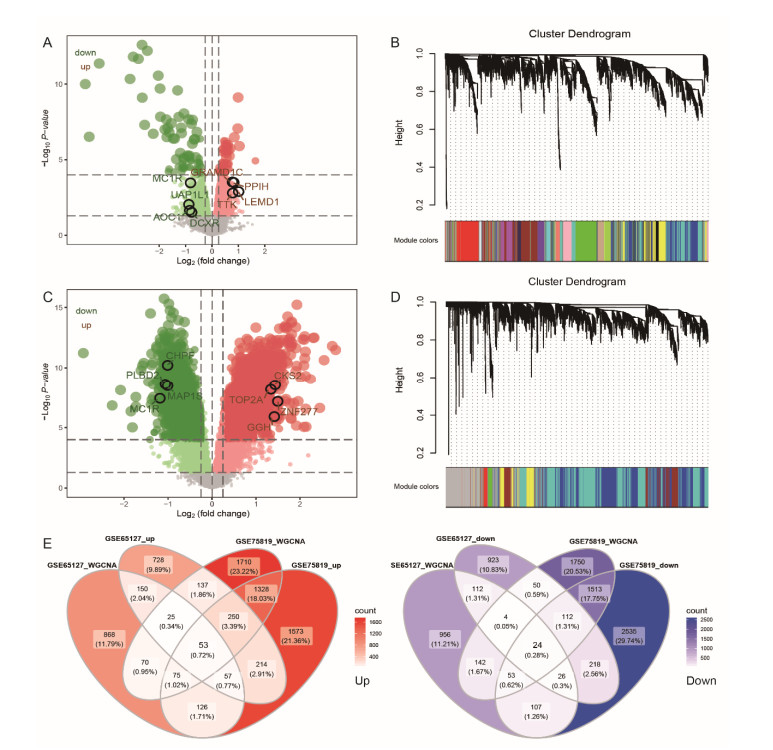
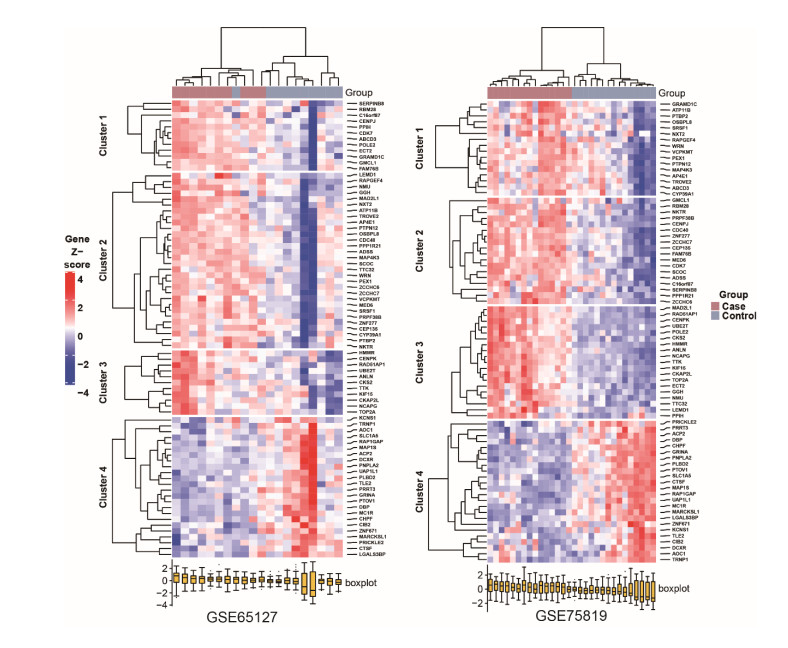
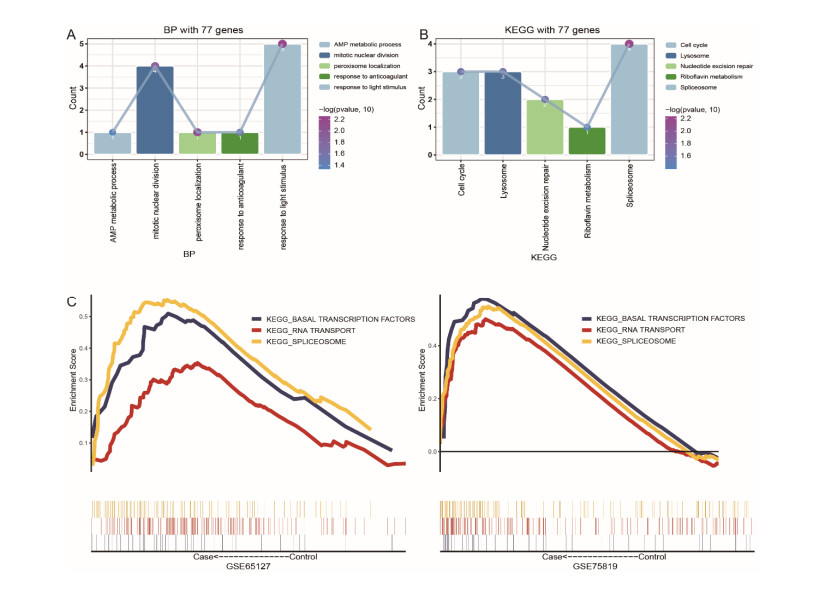
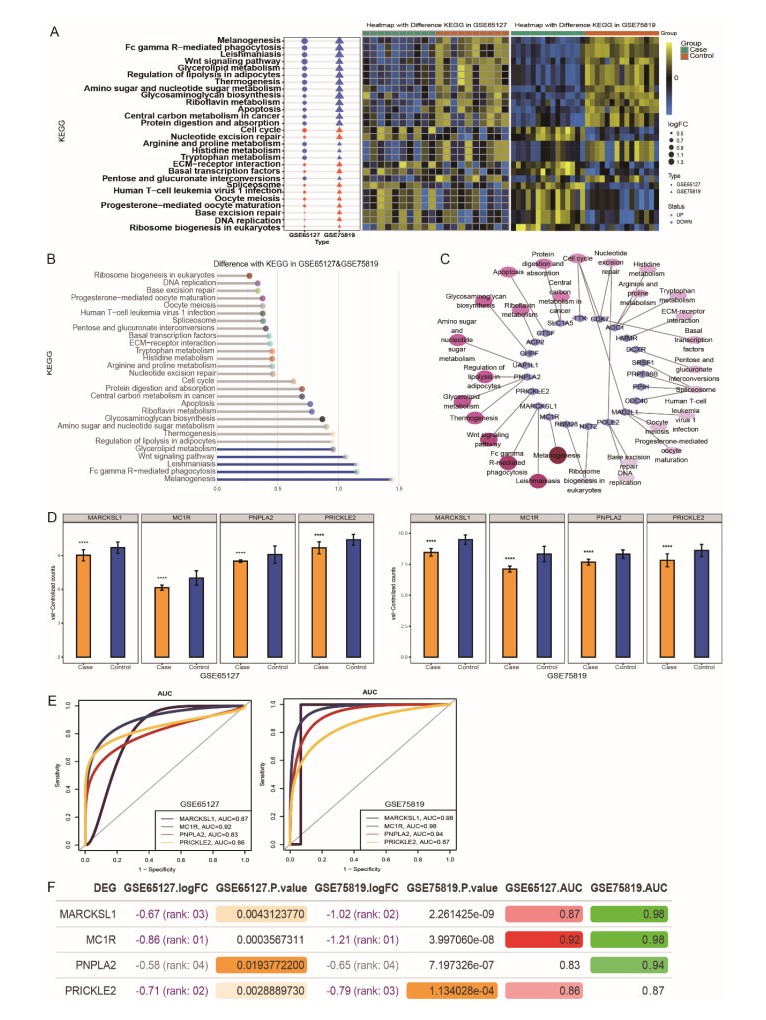
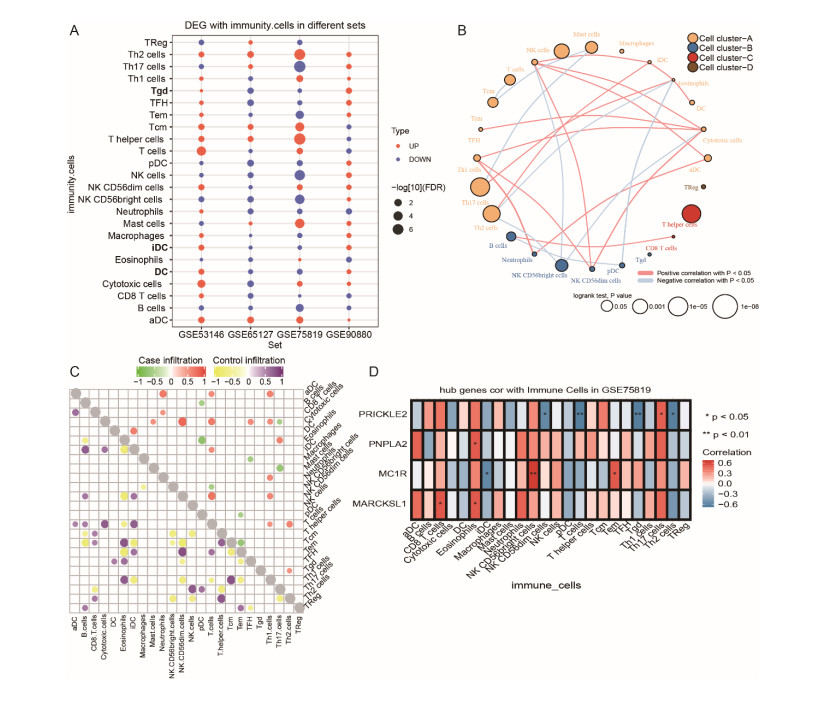
A total of 3083 DEGs and 16 modules were identified from GSE65127, and 5014 DEGs and 6 modules were screened from GSE75819. Finally, 77 important DEGs were identified. Enrichment analysis showed that 77 DEGs were mainly involved in spliceosome etc. The results of GSVA showed that melanogenesis, Fc gamma R-mediated phagocytosis, leishmaniasis, Wnt pathway and glycolipid metabolism were important KEGG pathways. The genes involved in these pathways were identified as key genes (MARCKSL1, MC1R, PNPLA2 and PRICKLE2). The AUC values of MC1R were the highest. Furthermore, different immune cells had different infiltration in vitiligo. There was a high correlation between immune cells and key genes.
MC1R was found as a key gene in vitiligo and involved in the melanogenesis. The immune cells were different infiltration in vitiligo. These results suggested that key genes may be used as markers of vitiligo, and were associated with immune cell, especially MC1R.
Citation: Xiangyue Zhang, Wen Hu, Zixian Lei, Hongjuan Wang, Xiaojing Kang. Identification of key genes and evaluation of immune cell infiltration in vitiligo[J]. Mathematical Biosciences and Engineering, 2021, 18(2): 1051-1062. doi: 10.3934/mbe.2021057
To improve the understanding of the molecular mechanism of vitiligo is necessary to predict and formulate new targeted gene therapy strategies.
GSE65127, GSE75819, GSE53146 and GSE90880 were collected, and obtained four groups of differentially expressed genes (DEGs) by limma R package. Through weighted gene co-expression network analysis (WGCNA), the co-expression of genes with large variance in GSE65127 and GSE75819 was identified. Enrichment analysis of intersection gene between module genes and DEGs with the same up-regulated or down-regulated in GSE65127 and GSE75819 was performed. In addition, ssGSEA was used to identify the immune infiltration of vitiligo in four datasets.
A total of 3083 DEGs and 16 modules were identified from GSE65127, and 5014 DEGs and 6 modules were screened from GSE75819. Finally, 77 important DEGs were identified. Enrichment analysis showed that 77 DEGs were mainly involved in spliceosome etc. The results of GSVA showed that melanogenesis, Fc gamma R-mediated phagocytosis, leishmaniasis, Wnt pathway and glycolipid metabolism were important KEGG pathways. The genes involved in these pathways were identified as key genes (MARCKSL1, MC1R, PNPLA2 and PRICKLE2). The AUC values of MC1R were the highest. Furthermore, different immune cells had different infiltration in vitiligo. There was a high correlation between immune cells and key genes.
MC1R was found as a key gene in vitiligo and involved in the melanogenesis. The immune cells were different infiltration in vitiligo. These results suggested that key genes may be used as markers of vitiligo, and were associated with immune cell, especially MC1R.

| [1] | A. Singh, V. Gotherwal, P. Junni, V. Vijayan, M. Tiwari, P. Ganju, et al., Mapping architectural and transcriptional alterations in non-lesional and lesional epidermis in vitiligo, Sci. Rep. , 7 (2017), 9860. |
| [2] | R. Dey-Rao, A. A. Sinha, Vitiligo blood transcriptomics provides new insights into disease mechanisms and identifies potential novel therapeutic targets, BMC Genomics , 18 (2017), 109. |
| [3] | V. N. Sehgal, G. Srivastava, Vitiligo: compendium of clinico-epidemiological features, Indian J. Dermatol. Venereol. Leprol. , 73 (2007), 149-156. |
| [4] | D. Chen, H. Tuan, E. Y. Zhou, D. Liu, Y. Zhao, D. Chen, et al., Quality of life of adult vitiligo patients using camouflage: A survey in a Chinese vitiligo community, PLoS One , 14 (2019), e0210581. |
| [5] | J. I. Silverberg, N.B. Silverberg, Vitiligo disease triggers: psychological stressors preceding the onset of diseas, Cutis , 95 (2015), 255-262. |
| [6] | A. Alkhateeb, P. R. Fain, A. Thody, A. Thody, D. C. Bennett, R. A. Spritz, Epidemiology of vitiligo and associated autoimmune diseases in Caucasian probands and their families, Pigm. Cell Res. , 16 (2003), 208-214. |
| [7] | S. W. Henning, D. Jaishankar, L.W. Barse, E. R. Dellacecca, N. Lancki, K. Webb, et al., The relationship between stress and vitiligo: Evaluating perceived stress and electronic medical record data, PLoS One , 15 (2020), e0227909. |
| [8] | P. E. Grimes, M. M. Miller, Vitiligo: Patient stories, self-esteem, and the psychological burden of disease, Int. J. Womens Dermatol. , 4 (2018), 32-37. |
| [9] | L. Raam, E. Kaleviste, M. Sunina, H. Vaher, M. Saare, E. Prans, et al., Lymphoid stress surveillance response contributes to vitiligo pathogenesis, Front. Immunol. , 9 (2018), 2707. |
| [10] | H. Xie, F. Zhou, L. Liu, G. Zhu, Q. Li, C. Li, et al., Vitiligo: How do oxidative stress-induced autoantigens trigger autoimmunity?, J. Dermatol. Sci. , 81 (2016), 3-9. |
| [11] | H. Atas, M. Gonul, Increased risk of metabolic syndrome in patients with vitiligo, Balk. Med. J. , 34 (2017), 219-225. |
| [12] | M. Rashighi, P. Agarwal, J. M. Richmond, T. H. Harris, K. Dresser, M. W. Su, et al., CXCL10 is critical for the progression and maintenance of depigmentation in a mouse model of vitiligo, Sci. Transl. Med. , 6 (2014), 223ra23. |
| [13] | J. Ocampo-Candiani, M. Salinas-Santander, V. Trevino, R. Ortiz-Lopez, J. Ocampo-Garza, C.N. Sanchez-Dominguez, Evaluation of skin expression profiles of patients with vitiligo treated with narrow-band UVB therapy by targeted RNA-seq, An. Bras. Dermatol. , 93 (2018), 843-851. |
| [14] | M. Rashighi, J. E. Harris, Vitiligo pathogenesis and emerging treatments, Dermatol. Clin. , 35 (2017), 257-265. |
| [15] | A. G. Tsadik, M. Z. Teklemedhin, T. Mehari Atey, M. T. Gidey, D. M. Desta, Public knowledge and attitudes towards vitiligo: A survey in Mekelle city, Northern Ethiopia, Dermatol. Res. Pract. , 2020 (2020), 3495165. |
| [16] | M. E. Ritchie, B. Phipson, D. Wu, Y. Hu, C. W. Law, W. Shi, et al., limma powers differential expression analyses for RNA-sequencing and microarray studies, Nucleic Acids Res. , 43 (2015), e47. |
| [17] | X. Shi, T. Huang, J. Wang, Y. Liang, C. Gu, Y. Xu, et al., Next-generation sequencing identifies novel genes with rare variants in total anomalous pulmonary venous connection, EBioMedicine , 38 (2018), 217-227. |
| [18] | L. Zhang, X. Shi, C. Gu, B. Chen, M. Wang, Y. Yu, et al., Identification of cell-to-cell interactions by ligand-receptor pairs in human fetal heart, Biochim. Biophys. Acta Mol. Basis Dis. , 1866 (2020), 165917. |
| [19] | P. Langfelder, S. Horvath, WGCNA: an R package for weighted correlation network analysis, BMC Bioinf. , 9 (2008), 559. |
| [20] | G. Yu, L. G. Wang, Y. Han, Q. Y. He, clusterProfiler: an R package for comparing biological themes among gene clusters, OMICS , 16 (2012), 284-287. |
| [21] | C. Gu, X. Shi, Z. Huang, J. Chen, J. Yang, J. Shi, et al., A comprehensive study of construction and analysis of competitive endogenous RNA networks in lung adenocarcinoma . Biochim. Biophys. Acta, Proteins Proteomics , 1868 (2020), 140444. |
| [22] | G. Bindea, B. Mlecnik, M. Tosolini, A. Kirilovsky, M. Waldner, A. C. Obenauf, et al., Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer, Immunity , 39 (2013), 782-795. |
| [23] | A. Taieb, Vitiligo as an inflammatory skin disorder: a therapeutic perspective, Pigment Cell Melanoma Res. , 25 (2012), 9-13. |
| [24] | J. C. Garcia-Borron, Z. Abdel-Malek, C. Jimenez-Cervantes, MC1R, the cAMP pathway, and the response to solar UV: extending the horizon beyond pigmentation, Pigment Cell Melanoma Res. , 27 (2014), 699-720. |
| [25] | E. I. Minder, J. Barman-Aksoezen, X. Schneider-Yin, Pharmacokinetics and pharmacodynamics of afamelanotide and its clinical use in treating dermatologic disorders, Clin. Pharmacokinet. , 56 (2017), 815-823. |
| [26] | X. Yuan, D. Meng, P. Cao, L. Sun, Y. Pang, Y. Li, et al., Identification of pathogenic genes and transcription factors in vitiligo, Dermatol. Ther. , 32 (2019), e13025. |
| [27] | N. A. Nagui, S. B. Mahmoud, R. M. Abdel Hay, M. M. Hassieb, L. A. Rashed, Assessment of gene expression levels of proopiomelanocortin (POMC) and melanocortin-1 receptor (MC1R) in vitiligo, Australas. J. Dermatol. , 58 (2017), e36-e39. |
| [28] | B. R. Kim, S. H. Lee, M. S. Park, S. H. Seo, Y. M. Park, Y. J. Kwon, et al., MARCKSL1 exhibits anti-angiogenic effects through suppression of VEGFR-2-dependent Akt/PDK-1/mTOR phosphorylation, Oncol. Rep. , 35 (2016), 1041-1048. |
| [29] | K. Aroni, S. Voudouris, E. Ioannidis, A. Grapsa, N. Kavantzas, E. Patsouris, Increased angiogenesis and mast cells in the centre compared to the periphery of vitiligo lesions, Arch. Dermatol. Res. , 302 (2010), 601-607. |
| [30] | P. Subramanian, S. P. Becerra, Role of the PNPLA2 gene in the regulation of oxidative stress damage of RPE, Adv. Exp. Med. Biol. , 1185 (2019), 377-382. |
| [31] | T. Cui, W. Zhang, S. Li, X. Chen, Y. Chang, X. Yi, et al., Oxidative stress-induced hmgb1 release frommelanocytes: a paracrine mechanism underlying the cutaneous inflammation in vitiligo, J. Invest. Dermatol. , 139 (2019), 2174-2184 e4. |
| [32] | T. Nagaoka, M. Furuse, T. Ohtsuka, K. Tsuchida, M. Kishi, Vangl2 interaction plays a role in the proteasomal degradation of Prickle2, Sci. Rep. , 9 (2019), 2912. |
| [33] | C. Niu, H. A. Aisa, Upregulation of melanogenesis and tyrosinase activity: potential agents for vitiligo, Molecules , 22 (2017), 1303. |
| [34] | S. Yu, C. E. Lan, H. S. Yu, Mechanisms of repigmentation induced by photobiomodulation therapy in vitiligo, Exp. Dermatol. , 28 (2019), 10-14. |
| [35] | T. Pillaiyar, M. Manickam, S.H. Jung, Downregulation of melanogenesis: drug discovery and therapeutic options, Drug Discov. Today , 22 (2017), 282-298. |
| [36] | S. Bournazos, J. V. Ravetch, Diversification of IgG effector functions, Int. Immunol. , 29 (2017), 303-310. |
| [37] | H. Weng, H. Huang, H. Wu, X. Qin, B. S. Zhao, L. Dong, et al., Mettl14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mrna m 6 a modification, Cell Stem Cell , 22 (2018), 191-205 e9. |
| [38] | C. Regazzetti, F. Joly, C. Marty, M. Rivier, B. Mehul, P. Reiniche, et al., Transcriptional analysis of vitiligo skin reveals the alteration of wnt pathway: a promising target for repigmenting vitiligo patients, J. Invest. Dermatol. , 135 (2015), 3105-3114. |
| [39] | R. Speeckaert, N. van Geel, Vitiligo: an update on pathophysiology and treatment options, Am. J. Clin. Dermatol. , 18 (2017), 733-744. |
| [40] | K. Boniface, A. Taieb, J. Seneschal, New insights into immune mechanisms of vitiligo, G Ital Dermatol. Venereol , 151 (2016), 44-54. |
 Supplementary-Table S1.pdf Supplementary-Table S1.pdf |
 |
| Supplementary-Table S2.pdf |
|
Figures(5)

Xiangyue Zhang, Wen Hu, Zixian Lei, Hongjuan Wang, Xiaojing Kang. Identification of key genes and evaluation of immune cell infiltration in vitiligo[J]. Mathematical Biosciences and Engineering, 2021, 18(2): 1051-1062. doi: 10.3934/mbe.2021057







 DownLoad:
DownLoad: